
Abstract
The continuously increasing incidence of diabetes worldwide has attracted the attention of the scientific community and driven the development of a novel class of antidiabetic drugs that can be safely and effectively used in diabetic patients. Of particular interest in this context are complications associated with diabetes, such as renal impairment, which is the main cause of high cardiovascular morbidity and mortality in diabetic patients. Intensive control of glucose levels and other risk factors associated with diabetes and metabolic syndrome provides the foundations for both preventing and treating diabetic nephropathy. Dipeptidyl peptidase-4 (DPP-4) inhibitors represent a highly promising novel class of oral agents used in the treatment of type 2 diabetes mellitus that may be successfully combined with currently available antidiabetic therapeutics in order to achieve blood glucose goals. Beyond glycemic control, emerging evidence suggests that DPP-4 inhibitors may have desirable off-target effects, including renoprotection. All type 2 diabetes mellitus patients with impaired renal function require dose adjustment of any DPP-4 inhibitor administered except for linagliptin, for which renal excretion is a minor elimination pathway. Thus, linagliptin is the drug most frequently chosen to treat type 2 diabetes mellitus patients with renal failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
1.1 Diabetic Nephropathy
Diabetes mellitus is defined as a metabolic disorder characterized by hyperglycemia that develops due to defects in insulin secretion, insulin action, or both [1]. Diabetes is a serious public health problem worldwide with increasing prevalence and incidence, resulting in increasing numbers of patients with diabetic complications [2]. In addition, approximately 193 million diabetic patients worldwide remain undiagnosed, which predisposes them to develop several chronic complications of untreated chronic hyperglycemia [3]. The harmful effects of chronic hyperglycemia are generally categorized into macrovascular complications (such as stroke and acute coronary syndromes) and microvascular complications (such as diabetic nephropathy, neuropathy, and retinopathy) [4]. Diabetic vascular complications are initiated through many metabolic and structural derangements, including the accumulation of advanced glycation end-products, abnormal stimulation of signaling cascades such as protein kinase C (PKC), overproduction of reactive oxygen species (ROS), and abnormal stimulation of hemodynamic regulation systems such as the renin–angiotensin–aldosterone system (RAAS) [5].
Diabetic kidney disease is one of the most common complications of both type 1 and type 2 diabetes mellitus (T1DM and T2DM, respectively), occurring in 20–30% of cases. The major consequences of diabetic nephropathy include loss of kidney function leading to end-stage renal disease (ESRD), accelerated cardiovascular disease, and death [6, 7]. However, much progress has been made in attempts to delay the progression of diabetic nephropathy, and ESRD in T2DM patients is largely preventable through the application of strict glycemic control and intensified antihypertensive treatment [8]. Considering the strong association between blood glucose levels and the risk of developing microvascular complications of diabetes such as diabetic nephropathy, the aim of treating diabetic patients should be to achieve and maintain a safe glucose concentration. In addition, treatment with angiotensin-converting enzyme (ACE) inhibitors has been shown to decrease the risk of developing nephropathy and cardiovascular events in patients with T2DM [4]. Despite the benefits derived from strict glycemic and blood pressure control, the incidence of these complications is still increasing [9]. Therefore, early diagnosis of diabetes and prompt administration of therapy (including diet and lifestyle modifications) will significantly reduce diabetes-associated comorbidities, including renal impairment, a complication which further influences therapeutic decision-making in this specific population.
1.2 The Influence of Renal Insufficiency on Drug Elimination and Drug-Metabolizing Enzyme Activity
Most drugs are eliminated via metabolism in the liver and/or excretion by the kidneys. Impaired renal function may considerably alter the pharmacokinetics and pharmacodynamics of drugs eliminated predominantly via renal excretory mechanisms [10]. Renal diseases affect glomerular blood flow, the plasma filtration rate, and tubular secretion and reabsorption, thus altering drug clearance. Renal metabolism may also be affected, and these are the two most common pharmacokinetics-related consequences of renal impairment [11]. There is increasing evidence that changes may be more prominent in T2DM patients with severely impaired renal function, and may be associated not only with the renal elimination of drugs but also with changes in drug absorption, hepatic metabolism, plasma protein binding, and drug distribution [10].
Two strategies are commonly used to optimize therapeutic outcomes and minimize the risk of drug toxicity in T2DM patients with impaired kidney function: firstly, medications that are not cleared by the kidney are chosen whenever possible; secondly, when the administration of drugs that are predominantly eliminated by the kidneys is unavoidable, dosing regimen adjustments are made according to guidelines, recommendations, or detailed pharmacokinetic calculations [12]. When necessary, dosing adjustments can be made either by reducing the drug dose or by extending the dosing interval [13]. A reasonable estimation of renal function can be gained from glomerular function calculations based on the calculated creatinine clearance (CLCR), which is in turn derived using the serum creatinine measured in a simple blood test. Given that drug clearance is proportional to CLCR, a practical method of calculating dose adjustments for kidney-cleared drugs is provided by measuring creatinine [14].
1.3 Objectives of the Review
This review focuses on the pharmacokinetic characteristics of the chemically heterogeneous class of DPP-4 inhibitors in T2DM patients with chronic kidney disease, especially their potentially renoprotective effects.
1.4 The Literature Search Strategy
For this purpose, an extensive search of original and review articles in the PubMed database that were published up to March 2019 was performed, using the following keywords: “type 2 diabetes mellitus,” “diabetic nephropathy,” “diabetic kidney disease,” “incretin,” “glucagon-like peptide-1,” “GLP-1,” “DPP-4,” and “dipeptidyl-peptidase 4 inhibitor.”
2 The Physiology of Incretins
The concept of incretins emerged following experiments conducted during the 1960s which proved that orally administered glucose induced a significantly increased insulin secretion response compared to intravenous administration, even though similar plasma glucose concentrations were achieved [15, 16]. This differential response to oral versus intravenously administered glucose, known as the incretin effect, is mediated by incretin peptides through the entero-insular axis. The incretin effect has been recognized as contributing 50–70% of the insulin secretion following oral administration of glucose in healthy human subjects [17]. Two primary intestinal hormones with incretin activity are glucose-dependent insulinotropic peptide (or gastric inhibitory peptide, GIP) and glucagon-like peptide-1 (GLP-1). They are secreted by the intestine in response to nutrient ingestion, and act as signaling messengers to regulate hormone secretion from pancreatic α and β cells in order to properly direct the disposal of nutrients [18].
GLP-1 is a product of the prohormone proglucagon, which is synthesized in intestinal enteroendocrine L cells. Both intestinal L cells and pancreatic α cells produce proglucagon, but the post-translational processing of the prohormone differs among these cell types. Proglucagon in pancreatic α cells is cleaved by prohormone convertase 2 into glucagon and major proglucagon fragments (MGF), rendering this molecule biologically inactive [19]. On the other hand, in intestinal L cells, proglucagon (“enteroglucagon”) is cleaved by prohormone convertase 1/3 into glicentin, oxyntomodulin, and two glucagon-like sequences, GLP-1 and GLP-2, which are released into the bloodstream. GLP-2 does not participate in glucose metabolism regulation, but it influences the growth and adaptation of the gut [20]. Additional post-translational modifications of GLP-1 involve the conversion of the carboxy-terminal amino acid glycine into an amide, which increases the stability of GLP-1 (1-36 amide) in plasma, as well as the hydrolysis of six carboxy-terminal amino acids, resulting in the 29-amino-acid peptide GLP-1 (7-36 amide), the major form of GLP-1 with intrinsic incretin activity [21].
GLP-1 secretion was initially described in response to glucose ingestion; however, ingestion of a mixed meal results in a more intense GLP-1 response than glucose alone. Additionally, bile acids released from the gallbladder postprandially have been shown to induce GLP-1 synthesis and secretion through the activation of Takeda G protein-coupled receptor 5 (TGR5) [22, 23].
The GLP-1 level in plasma rises postprandially. The response begins within ten minutes during the cephalic phase of a meal, with the peak concentration (40–80 pM) reached after an hour in healthy individuals, which should be compared with fasting concentrations (5–15 pM) that are secreted at low tonic rates [24]. However, GLP-1 is a short-acting molecule due to rapid and extensive degradation in the bloodstream by the ubiquitous enzyme DPP-4, with a half-life of 1.5–3 min [25]. DPP-4 cleaves two amino-terminal residues, hindering the biological activity of GLP-1. Inactivated GLP-1 (9-36 amide) is further cleaved in the kidneys by DPP-4, which is expressed on the apical surfaces of endothelial cells of the renal capillaries and the brush-border membranes of tubular cells [26].
The main action of GLP-1 is the glucose-dependent stimulation of insulin biosynthesis and secretion. The action of GLP-1 in β cells is self-limiting and prevents the development of hypoglycemic events. When a reduction in plasma glucose levels is achieved by the secretion of insulin, GLP-1 signaling is also inhibited [27]. GLP-1 stimulates glucose-dependent insulin secretion and inhibits glucagon secretion, thus preventing the development of hyperglycemia (in contrast to sulfonylurea, GLP-1 has no effect on insulin release when the glycemic level is below 4 mM). GLP-1 activation has also been shown to inhibit the apoptosis of β cells and to enhance β-cell proliferation as well as β-cell neogenesis from ductal precursor cells [28, 29]. In addition to pancreatic islets, the expression of the GLP-1 receptor has been detected in various cell and tissue types, including the central nervous system (CNS), heart, lung, blood vessels, gastrointestinal tract, liver, kidney, muscle, and adipose tissue [30]. This wide distribution of the GLP-1 receptor could explain the observed extrapancreatic effects of GLP-1. GLP-1 exerts effects on gastrointestinal motility and secretion (delaying gastric emptying), inhibits appetite, and reduces food intake; taken together, these effects lead to a reduction in body weight [31].
DPP-4, also known as the T-cell activation antigen CD26 (cluster of differentiation 26), is a membrane-associated peptidase composed of 766 amino acid residues. It is widely distributed in different cell and tissue types, including T and B lymphocytes, vascular endothelium, kidneys, hepatocytes, and the intestinal brush-border membrane [32]. DPP-4 is a 110-kDa type II membrane-spanning protein that is involved in signal transduction, as it activates a network of intracellular signaling pathways [33]. Membrane-bound DPP-4 manifests numerous nonenzymatic activities upon colocalizing with other membrane proteins and modulating their intrinsic activity. Among these proteins, DPP-4 interacts with adenosine deaminase, caveolin-1, insulin-like growth factor 2 receptor (IGF2R), and glypican-3 [34]. DPP-4 also exists as a soluble circulating isoform in plasma, originating from transmembrane and intracellular domains [35]. Therefore, biological effects of DPP-4 include enzymatic as well as signaling functions. Soluble DPP-4 is an exopeptidase which hydrolytically cleaves peptides (including GLP-1) at the second amino acid position from the amino-terminal end of the molecule, especially if the second amino acid is alanine or proline [36]. In addition to GLP-1, DPP-4 plays a significant role in the metabolism of neuropeptide Y, which is involved in the regulation of food intake and obesity. DPP-4 also exerts numerous effects on metabolic regulation via other regulatory peptides, including GIP, insulin-like growth factor 1 (IGF-1), gastrin-releasing peptide, endomorphin, and enterostatin [36].
GLP-1, a molecule that has a high potency to stimulate insulin biosynthesis and secretion and has highly desirable effects on satiety and food intake, degrades rapidly. This has attracted the attention of the scientific community and has driven the development of hydrolysis-resistant GLP-1 receptor agonists and inhibitors of DPP-4 as novel therapeutic approaches that aim to restore GLP-1 signaling and insulin secretion in patients with T2DM.
3 Incretin-Based Therapy of T2DM
Several GLP-1 receptor agonists have gained regulatory approval for T2DM treatment. Exenatide is a full and equipotent GLP-1 receptor agonist that is not degraded by DPP-4 and is more slowly eliminated than endogenous GLP-1. Exenatide was the first clinically approved short-acting GLP-1 receptor agonist. It is administered subcutaneously by injection twice a day or is available in a prolonged-release formulation that is suitable for once-a-week administration [37]. Liraglutide is a GLP-1 receptor agonist that is obtained by attaching a palmitoyl residue to the GLP-1 molecule. It can therefore be transported through the blood while bound to albumin, resulting in DPP-4 resistance and a half-life of 13 h [38]. Novel agents have subsequently been introduced, including once-a-day lixisenatide and once-weekly exenatide, dulaglutide, and semaglutide [39, 40]. GLP-1 receptor agonists have mild and transient gastrointestinal side effects—mainly nausea and vomiting—due to the resulting delayed gastric emptying. The main disadvantage of GLP-1 receptor agonists is the route of administration (subcutaneous injection).
3.1 DPP-4 Inhibitors
Following the discovery of GLP-1, the inhibition of DPP-4 became a major research target, since the inhibition of DPP-4 has a profound influence on incretin hormone activity by increasing the plasma concentrations of endogenous and active peptides [41]. DPP-4 inhibitors or so-called gliptins were originally developed as anti-immune therapies, because DPP-4 is expressed as the T-cell activation antigen CD26 by certain immune cells [33]. Early animal studies demonstrated that DPP-4 inhibition can hinder rapid incretin degradation, increasing the postprandial levels of GLP-1 and reducing plasma glucose levels postprandially due to the stimulation of insulin secretion and the reduction of liver glucose production [42]. The majority of the effects related to the inhibition of DPP-4 are ascribed to increased GLP-1 levels. Therefore, DPP-4 became a significant target for the treatment of T2DM. DPP-4 inhibitors reduce DPP-4 activity by 70–90%, they do not pass through the blood–brain barrier, and they do not exert direct effects on satiety or gastric emptying [43].
Sitagliptin was the first DPP-4 inhibitor to be approved in the USA (in 2006) and in Europe (in 2007) for T2DM treatment [44]. So far, five gliptins have gained approval for clinical use: sitagliptin, vildagliptin, saxagliptin, linagliptin, and alogliptin. Three others—teneligliptin, anagliptin, and trelagliptin—are approved only for the Japanese and Korean markets [43]. Even though all gliptins rely on the same principle of action, differences in pharmacokinetic properties may impact clinical decision-making. Gliptins have been classified into three classes according to the binding mode at the active center of DPP-4 [45]. Vildagliptin and saxagliptin, which are class one DPP-4 inhibitors, are competitive reversible DPP-4 inhibitors which bind to the S1 and S2 subsites of an enzyme and form covalent bonds with Ser630 within the active site of DPP-4. Alogliptin and linagliptin are class two inhibitors, whereas sitagliptin and teneligliptin are class three gliptins [46]. Alogliptin and linagliptin bind to the S1’ and S2’ subsites of the enzyme, respectively, inducing a conformational change in Tyr547 at the S1′ subsite, which results in increased inhibitory potency. The interaction of linagliptin with the S1’ subsite results in an eightfold increase in activity compared to alogliptin. The third class of gliptins, sitagliptin and teneligliptin, have the highest inhibitory potencies for DPP-4, since both of these drugs interact with the S2-extensive subsite of the DPP-4 active center. Therefore, the increased potencies of these gliptins appear to result from an increase in the number of interactions with the enzyme DPP-4 [43, 45]. In addition to lowering glycemia, sitagliptin has been shown to reduce the free fatty acid level in blood and to exert anti-inflammatory effects, meaning that it has insulin-sensitizing properties [47]. In T2DM patients with renal impairment, sitagliptin has more favorable effects than sulfonylureas [48]. Anagliptin, which has thus far only gained regulatory approval in Japan (in 2012), has been shown to exert beneficial cardiovascular effects through its lipid-lowering and antiatherogenic activities [49]. Anagliptin is excreted via the kidneys, and renoprotective effects of this gliptin have been demonstrated in T2DM patients with diabetic nephropathy [50]. Anagliptin also has a stronger anti-inflammatory action than sitagliptin [51]. Additionally, numerous reports on the effects of gliptins on the serum lipid profile, inflammation, and blood pressure may explain the potential cardioprotective effects of this class of antidiabetics [52].
Renal excretion is the predominant elimination pathway for sitagliptin and alogliptin, whereas hepatic metabolism is also important for saxagliptin (this leads to an active metabolite with approximately half the biological potency of saxagliptin). On the other hand, linagliptin is mainly excreted by the biliary route, so T2DM patients with impaired renal function who are taking this drug do not require dose adjustment [53]. The main pharmacokinetic parameters of DPP-4 inhibitors in healthy volunteers are shown in Table 1.
DPP-4 inhibitors are available as oral drug formulations, and have mild side-effect profiles. The side effects of DPP-4 inhibitors include minor gastrointestinal side effects, skin rash, and nasopharyngitis, whereas hypoglycemia is uncommon. Also, this class of drugs has no effect on body weight [27]. Many are available in fixed combinations with metformin, including a sustained release formulation. Combination therapy with DPP-4 inhibitors and insulin results in a reduction in the insulin dose required to lower glycemic levels, an effect associated with a reduced incidence of hypoglycemic events [27].
Whereas DPP-4 inhibitors increase GLP-1 two- to threefold, GLP-1 receptor agonists increase GLP-1 up to tenfold, and are superior to DPP-4 inhibitors in terms of insulin secretion, reducing HbA1c levels, and body weight effects. However, GLP-1 agonists should be avoided in T2DM patients with GFR < 30 mL/min. Metformin is also contraindicated in T2DM patients with impaired renal function (due to the induction of lactic acidosis), while sodium-glucose cotransporter-2 (SGLT-2) inhibitors should not be used in T2DM patients with GFR < 45 mL/kg. In addition, hypoglycemic events that may occur during sulfonylurea treatment can be avoided by administering DPP-4 inhibitors, meaning that these agents are preferred in T2DM patients with impaired renal function [54].
4 The Entero-Renal Axis
Incretin-based therapy exerts numerous regulatory effects and benefits extending beyond glucose homeostasis. The entero-endocrine system has emerged as an important contributor to the regulation of water and electrolyte homeostasis upon meal ingestion. These effects are mediated by adjusting thirst via cerebral osmoreceptors located in the vascular organ of the forebrain (lamina terminalis), by controlling the intestinal transport of water and electrolyte absorption and secretion, through the appropriate disposal of absorbed water and nutrients to intra- and extracellular compartments, and by regulating renal secretion and the reabsorption of water and electrolytes, implying that it operates in a very flexible manner [53, 55]. Given that renal homeostasis of electrolytes is regulated slowly by circulating hormones and the diurnal rhythm, a speculated fast-acting entero-renal axis could contribute by swiftly responding to acute solute ingestion. For example, an increased urinary excretion of excess potassium was observed after a potassium-rich meal [56]. Results of another study demonstrated that an equivalent sodium load is excreted in the urine more rapidly after ingestion than following intravenous administration, independent of the effects of aldosterone and atrial natriuretic peptide levels, lending support to this theory [57]. In addition to the sodium balance, an enteral-assisted feed-forward loop for potassium and phosphate homeostasis has been reported [56, 57]. The intestine presumably senses changes in the concentrations of ingested electrolytes and responds by releasing hormone effectors of the entero-renal axis, such as GLP-1, peptide YY, ghrelin, secretin, guanylin, and vasoactive intestinal peptide (VIP), and by activating neural pathways to regulate the gastrointestinal and renal tubular transport processes [58]. Impairment of entero-renal axis signaling could be considered a pathophysiological element of salt-sensitive hypertension due to reduced sodium excretion.
GLP-1 receptors are expressed in the renal proximal tubular cells as well as in renal blood vessels [59, 60]. The participation of GLP-1 in the entero-renal axis was described following findings that exenatide infusion in healthy volunteers as well as in T2DM patients or liraglutide administration in T2DM patients increased natriuresis and thus played a prominent role in the control of sodium excretion [61, 62]. The underlying mechanism for this effect is the downregulation of sodium hydrogen exchanger 3 (NHE3) function, with a consequent decline in sodium reabsorption [63]. In accordance, DPP-4 has been shown to coexist in physical complexes with NHE3 in the brush border of proximal tubules, and it was found that DPP-4 inhibition also downregulates apical sodium–hydrogen exchange [64].
In further support, an acute oral load of sodium chloride, as well as a water load, resulted in increased circulating GLP-1 within five minutes [65, 66]. Also, GLP-1 receptor agonist administration has been shown to reduce water and saline intake independently of food intake, whereas administration of exendin-9, a GLP-1 receptor blocker, resulted in a dipsogenic effect, implying that endogenous GLP-1 tone suppresses drinking behavior, which could be an effect of GLP-1-producing cells in the nucleus tractus solitarii [67,68,69]. Furthermore, infusion of GLP-1 in healthy males reduced water intake following a salty meal without fluctuations in the serum concentration of sodium [68].
Even though intestinal hormones have the propensity to modulate tubular electrolyte handling without significantly varying the filtered load, signals from the gastrointestinal tract affecting postprandial renal blood flow can also aid in renal solute excretion. Ingestion of a high-protein meal has been shown to increase the glomerular filtration rate and to enhance the filtration of circulating solutes, independently of fluctuations in arterial pressure [70]. Thus, a postprandial increase in glomerular filtration ameliorates urinary excretion of nitrogen waste products and other metabolites that require renal excretion, making it an effective mechanism for the elimination of potentially harmful catabolic products and absorbed solutes.
5 Effects of DPP-4 Inhibitors on Renal Function and Renal Risk Factors
In recent years, three new classes of antihyperglycemic drugs with determined renoprotective potential have been successfully introduced for the treatment of T2DM: DPP-4 inhibitors, GLP-1 receptor agonists, and SGLT-2 inhibitors. Emerging evidence suggests that DPP-4 inhibitors used to treat T2DM may have renoprotective effects beyond glycemic control [53].
The main pharmacological effects of DPP-4 inhibitors were demonstrated to be the result of the accumulation of incretin hormones and the enhancement of their actions; in particular those of GLP-1, which exerts its effects via a specific GLP-1 receptor. However, DPP-4 cleaves multiple substrates other than GLP-1, which implies that pleiotropic actions of DPP-4 inhibitors may occur through incretin-independent mechanisms [71].
In rats, exogenous GLP-1 dose-dependently increased diuresis and natriuresis [72]. That initial evidence of the effects of a gut hormone, GLP-1, on renal function in rats was further confirmed in preclinical and clinical studies. The diuretic and natriuretic response to the intravenous infusion of recombinant GLP-1 was demonstrated in rats. GLP-1 was shown to inhibit sodium reabsorption in the proximal tubule, and, to a lesser extent, to increase the renal blood flow and/or glomerular filtration rate [73]. Potential renoprotective activity of GLP-1 at the level of the proximal renal tubule was observed in healthy subjects and in insulin-resistant obese men. More specifically, it was demonstrated that intravenous infusion of GLP-1 enhances sodium excretion, decreases hydrogen secretion, and ameliorates glomerular hyperfiltration in obese men [61].
The main mechanism responsible for the enhancement of diuresis and natriuresis seems to involve the inhibition of NHE3. As previously stated, DPP-4 is abundantly expressed in the kidney, with the highest level of expression occurring at the brush border of the S1–S3 segment of the proximal tubule. It has been shown that DPP-4 coimmunoprecipitates and redistributes with NHE3 to the microvilli-enriched fractions, whereas the binding of angiotensin II (Ang II) to the AT1 receptor in the proximal tubule stimulates NHE3 activity through multiple signaling pathways [64, 74, 75]. In accordance, the administration of angiotensin system inhibitors and DPP-4 inhibition may offer a new therapeutic approach that involves (at the very least) reducing sodium reabsorption in patients with diabetic nephropathy [76, 77].
The systemic infusion of GLP-1 increased the glomerular filtration rate and reduced proximal tubular sodium, bicarbonate, and water reabsorption in rats. This effect was suggested to be mediated, at least in part, by NHE3 inhibition as a consequence of the activation of the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) signaling pathway. Subsequent to activation, phosphorylation of PKA consensus sites occurs at the C-terminal region of NHE3 at serine residues 552 and 605 in brush-border microvilli membrane vesicles [78]. The same cascade may be involved in the regulation of vascular renal resistance, leading to increases in renal blood flow and the glomerular filtration rate [79]. These results suggesting that endogenous GLP-1 receptor signaling exerts a tonic natriuretic action via NHE3 inhibition were further confirmed by a study using the GLP-1 receptor antagonist exendin-9 in rats. The acute renal effects of GLP-1 receptor blockade were evaluated, and antidiuretic and antinatriuretic effects were observed in normoglycemic rats. These were accompanied by the stimulation of proximal tubule sodium reabsorption due to reduced PKA-mediated inhibition of NHE3 transport function [80].
Furthermore, GLP-1 was shown to exert renoprotective effects by inhibiting ATII signaling and proinflammatory action in glomerular endothelial cells. Namely, it was demonstrated in vivo and in vitro that hyperglycemia can activate PKC-β isoforms, which enhance ATII-associated detrimental effects in glomerular endothelial cells through increased degradation of GLP-1 receptors. Therefore, it was suggested that therapeutic strategies which aim to increase GLP-1 receptor signaling should be used to prevent glomerular endothelial dysfunction and the progression of diabetic nephropathy [81].
DPP-4 inhibitors were shown to exert natriuretic effects in a number of preclinical studies. Two specific inhibitors of DPP-4 (diprotin A and P32/98) significantly decreased NHE3 activity in opossum kidney proximal (OKP) tubule cells as a result of tyrosine kinase signaling pathway inhibition rather than by PKA activation [64]. The inhibition of NHE3 may result from the renal accumulation of GLP-1 due to DPP-4 inhibition. On the other hand, it was shown that the natriuretic activity of DPP-4 inhibitors may, in part, occur via mechanisms that do not involve GLP-1 and its receptor. Specifically, the DPP-4 inhibitor alogliptin also induced diuresis and natriuresis in mice lacking the GLP-1 receptor, in contrast to exendin-4, an agonist of the GLP-1 receptor. These results provided the initial evidence that DPP-4 may regulate multiple peptide substrates other than GLP-1 and affect the renal reabsorption of water and sodium [82]. Some of those substrates of DPP-4 include brain natriuretic peptide (BNP), substance P, neuropeptide Y (NPY), stromal cell-derived factor 1α (SDF-1α), and high-mobility group protein B1 (HMGB1), which are involved in vascular tone regulation, inflammation, cell migration, and cell differentiation [71]. Therefore, the off-target effects of DPP-4 inhibitors may be a consequence of interactions with these biologically active peptides.
SDF-1 was suggested to partly mediate the DPP-4 inhibitor-induced natriuretic response. SDF-1 and its cognate receptor, CXCR4, are expressed in both cortical and medullary tissues of kidneys in mice, predominantly in the SDF-1α isoform. SDF-1 regulates urinary sodium excretion in the distal nephron, presumably via the Na+/Cl− cotransporter or the epithelial sodium channel, which impacts renal hemodynamics [83].
Renoprotective effects of DPP-4 inhibitors have been confirmed in preclinical studies involving diabetic kidney disease models as well as other progressive kidney disease models. As previously stated, kidneys contain high levels of DPP-4. Furthermore, an increase in DPP-4 levels was reported in kidneys of rats fed a high-fat diet and those treated with streptozotocin [84]. This model was shown to be a suitable animal model for studying the final stages of T2DM, characterized by insulin deficiency [85]. Since both a high-fat diet and insulin deficiency are closely linked with the etiology of T2DM, a role of DPP-4 inhibitors in the treatment of diabetic nephropathy was suggested [84]. However, DPP-4 inhibitors are a chemically heterogeneous class of drugs that show significant pharmacological differences. Major attention has been focused on linagliptin, since it is the most potent DPP-4 inhibitor and the only compound that is not predominantly excreted in the urine and thus does not require dose adjustment in T2DM patients with renal impairment [86]. It also has the largest volume of distribution, which may enable good penetration in renal tissues. The ability of linagliptin to penetrate into kidney tissue was confirmed in wild-type and DPP-4-deficient rats using whole-body autoradiography [87].
Linagliptin was shown to exert renoprotective effects in several animal models of diabetic nephropathy. It reduced albuminuria in a model of early diabetic nephropathy of streptozotocin-induced diabetes in rats, and attenuated the expression of the oxidative stress markers NADPH oxidase (NOX) 2 and 4 [86, 88]. Previously, linagliptin, when used in combination with the angiotensin receptor blocker telmisartan, was found to reduce urinary albumin excretion and kidney malondialdehyde immunoreactivity, a marker of oxidative stress, in diabetic endothelial nitric oxide synthase (eNOS) knockout mice [77]. In addition to upregulating renal SDF-1 expression, linagliptin suppressed the progression of glomerular sclerosis, periglomerular fibrosis, podocyte loss, and renal oxidative stress in diabetes-prone mice lacking the GLP-1 receptor. This implies that DPP-4 inhibition, independent of GLP-1 receptor signaling, protects against diabetic nephropathy through SDF-1-dependent antioxidative and antifibrotic effects [83]. In addition, antifibrotic effects of linagliptin were reported in a mouse model of diabetic nephropathy characterized by extensive fibrosis. Linagliptin treatment of streptozotocin-induced diabetic mice ameliorated kidney fibrosis, regardless of the blood glucose level, by suppressing the activated fibroblast-generating endothelial-to-mesenchymal transition (EMT) pathway, at least in part [89].
Renoprotective properties have been determined for other DPP-4 inhibitors in animal models of diabetic kidney disease. In an animal model of obese T2DM, sitagliptin prevented β-cell dysfunction and evolution of pancreatic damage. Sitagliptin improved the metabolic profile in Zucker diabetic fatty (ZDF) rats through the amelioration of glucose and triglyceride levels and insulin resistance. It was also found to exert antiapoptotic and anti-inflammatory effects, since it reduced the Bax/Bcl-2 ratio and completely prevented the increased pancreatic overexpression of IL-1β and TRIB3 found in diabetic animals [90]. Furthermore, the DPP-4 inhibitor vildagliptin was shown to significantly decrease albuminuria and the urinary albumin/creatinine ratio, improve creatinine clearance, and inhibit interstitial expansion and glomerulosclerosis in a rat model of streptozotocin-induced T1DM. The renoprotective activity of vildagliptin was confirmed by a reduction in transforming growth factor beta 1 (TGF-β1) overexpression, since antifibrotic changes in the kidneys occur together with inhibition of the EMT that is usually initiated by TGF-β1 [91].
According to published studies, DPP-4 inhibitors may affect renal function not only directly but also through the modulation of renal risk factors, including oxidative stress, inflammation, and fibrosis. DPP-4 inhibitors mitigated endothelial dysfunction and reduced inflammation, thus providing efficient cardiovascular and renal protection [92]. The mechanisms for the anti-inflammatory activity of DPP-4 inhibitors in cardiovascular and renal protection include their impacts on pro- and anti-inflammatory mediators, cytokines, chemokines, immune cells, cell adhesion molecules, enzymes, transcription factors, pro- and antiapoptotic mediators, and oxidative stress markers. These protective properties of DPP-4 inhibition may be dependent on or independent of GLP-1, and they may rely on many factors such as the specific types of cell and tissue, the regional expression of cytokines and chemokines, and the type of disorder [93].
Glucose-independent effects of DPP-4 inhibitors on dyslipidemia, obesity, and blood pressure as renal risk factors in T2DM were also investigated. While GLP-1 receptor agonists were shown to reduce the levels of total cholesterol, LDL cholesterol, and triglycerides [94], DPP-4 inhibitors were suggested to have only a modest effect on total cholesterol levels [95]. Similarly, the results of a meta-analysis demonstrated that DPP-4 inhibitors, in contrast to GLP-1 receptor agonists, do not affect body weight in patients with T2DM [96]. This discrepancy between the results obtained using GLP-1 receptor agonists and those attained with DPP-4 inhibitors indicates that there are GLP-1-independent mechanisms of DPP-4 inhibitors. In patients with T2DM, it was shown that sitagliptin increases the level of total peptide YY (PYY). PYY, a DPP-4 substrate, is one of the physiological regulators of food intake [97]. On the other hand, DPP-4 inhibitors may exert modest blood-pressure-lowering effects in patients with T2DM, independent of weight loss [98]. Mechanisms linking sustained GLP-1 receptor signaling to blood pressure reductions are yet to be fully understood, but may include contributions from natriuresis and diuresis, improved vasorelaxation, endothelial function, and neurohormonal pathways [53].
Despite numerous results demonstrating renoprotective effects of DPP-4 inhibitors in animal models of diabetic and nondiabetic nephropathy, clinical evidence confirming renal benefits is scarce. The most recent and significant clinical study investigating the impact of DPP-4 inhibitors on renal function in patients with T2DM is the CARMELINA (Cardiovascular and Renal Microvascular Outcome Study with Linagliptin) study. In a large patient population at very high cardiovascular and renal risk, i.e., in patients with T2DM and concomitant atherosclerotic vascular disease and/or diabetic kidney disease, the DPP-4 inhibitor linagliptin did not modulate risk for heart-failure hospitalization or other associated heart-failure-related complications [99, 100]. This finding is particularly relevant in view of results of previous randomized clinical studies with saxagliptin (SAVOR-TIMI 53) [101] and alogliptin (EXAMINE) [102] that reported significant increases in this outcome, whereas sitagliptin (TECOS) [103] showed no effect on the heart failure hospitalization rate. These results clearly illustrate the heterogeneity across the DPP-4 inhibitor class with regard to effects on the risk for heart failure outcomes, with linagliptin and sitagliptin having no effect and saxagliptin increasing this risk. Molecular mechanisms responsible for these pharmacological variations are still to be elucidated, but the differences in their molecular structures may, at least in part, account for diverse off-target effects [99].
6 Pharmacokinetics of DPP-4 Inhibitors and Dose Adjustment in T2DM Patients with Renal Impairment
Sitagliptin was the first agent in the DPP-4 inhibitor class to gain FDA approval in the USA, where it has been sold under the tradename Januvia since October 2006. Sitagliptin is an oral, once-daily DPP-4 inhibitor that is available as 25-mg, 50-mg, and 100-mg tablets. It has been used as monotherapy and in combination with other antidiabetic drugs to improve glycemic control. A sitagliptin/metformin fixed combination (Janumet, Merck) in tablet form was approved in April 2007. Sitagliptin demonstrates 87% bioavailability of the oral dose [104]. Peak plasma levels of sitagliptin occur approximately 1–4 hours after oral administration. The apparent terminal half-life of sitagliptin is approximately 10–12 h. In pharmacokinetic studies of various dosages of sitagliptin, the maximal concentration (Cmax) and the area under the plasma concentration–time curve (AUC) increased in a dose-proportional manner. This drug demonstrates low binding to serum proteins. Sitagliptin does not interfere with cytochrome P450 (CYP) enzymes. In healthy subjects, 75–80% of the drug is excreted unchanged in the urine. The main mechanisms of elimination are active secretion and glomerular filtration. Metabolites were detected at low levels, although these are not expected to improve the DPP-4-inhibitory effect of sitagliptin [105,106,107].
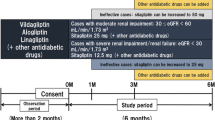
Single-dose, open-label studies were conducted to evaluate the effect of renal insufficiency on the pharmacokinetics of sitagliptin in T2DM patients. Patients with mild renal insufficiency (CLCR ≥ 50 mL/min) exhibited a less than twofold increase in plasma AUC levels compared with control patients who had normal renal function. In T2DM patients with moderate (CLCR 30–50 mL/min) and severe (CLCR < 30 mL/min) renal impairment, including those on hemodialysis, plasma sitagliptin concentrations were approximately 2.3–4.5 times higher than in subjects with normal or mildly impaired renal function. This also corresponded to a Cmax increase of 1.4-fold to 1.8-fold and an increased terminal half-life of up to 22.5 h with severe renal impairment compared to values in subjects with normal renal function (10–12 h). During hemodialysis, 13.5% of the sitagliptin was removed over a 3- to 4-hour session starting 4 h after oral administration [108, 109]. Therefore, in T2DM patients with renal impairment, it is recommended that the dose should be reduced from 100 mg to 50 mg daily if GFR is 30–50 mL/min, and to 25 mg daily if GFR is less than 30 mL/min. For T2DM patients on hemodialysis or peritoneal dialysis, it is recommended that the dose should not exceed 25 mg daily [110].
Saxagliptin, approved in July 2009, is usually administered once daily at a therapeutic dose of 5 mg. Saxagliptin is well absorbed orally and food does not have a clinically relevant impact on its pharmacokinetics. Peak levels of saxagliptin occur within 2 hours. Although the overall half-life is short, the metabolism of saxagliptin, which is mediated primarily by the CYP3A4/5 system, produces an active metabolite (5-hydroxy saxagliptin) that retains 50% of the hypoglycemic activity of the parent drug [105]. Therefore, inhibitors and inducers of this system are expected to affect the concentration of saxagliptin. However, clinically relevant drug–drug interactions have not been detected in combinations with CYP inhibitors or inducers when used at a dose of 2.5 mg once daily. This drug shows weak binding to proteins in the serum. 75% of the administered dose of saxagliptin, including 5-hydroxy saxagliptin and other minor hydroxylated inactive metabolites, was eliminated via the renal route and recovered in urine [105, 111].
Single-dose, open-label studies were conducted to evaluate the effect of renal failure on the pharmacokinetics and tolerability of saxagliptin and its pharmacologically active metabolite, 5-hydroxy saxagliptin, in nondiabetic subjects. No clinically significant differences in saxagliptin or 5-hydroxy saxagliptin pharmacokinetics were observed in subjects with mild renal impairment. However, in T2DM patients with moderate or severe renal impairment, dose reduction is recommended due to higher systemic exposure to saxagliptin and its metabolite (expressed as the AUC) [111, 112]. Therefore, in order to maintain plasma levels comparable to those in T2DM patients with normal renal function, the starting dose of saxagliptin should be reduced by 50% (2.5 mg/daily) in T2DM patients with moderate or severe renal impairment or ESRD, but no dose adjustment is recommended for those with mild renal impairment [110].
Vildagliptin is an orally active, potent, and selective DPP-4 inhibitor that is used either as monotherapy or in combination with a variety of other antidiabetic drugs. It is administered at a therapeutic dose of 50 mg twice daily. Following oral administration, vildagliptin is rapidly and well absorbed. Its absolute oral bioavailability is approximately 85%. A dose-dependent increase in exposure to vildagliptin (expressed as the AUC) over the dose range 25–200 mg has been documented. Food does not affect its pharmacokinetics. Vildagliptin is minimally bound to plasma proteins (9.3%). Based on its volume of distribution (71 L), it is assumed to be distributed extensively in the extravascular compartment [113]. The primary metabolic pathway is cyano-group hydrolysis by various organs with little CYP involvement. Minor metabolites are formed by amide bond hydrolysis, glucuronidation, or oxidation at the pyrrolidine moiety of vildagliptin. Due to minimal involvement of CYP enzymes in the overall metabolism, vildagliptin has a low potential for drug interactions. The major circulating components in the plasma are the unchanged drug and a major metabolite generated by hydrolysis, LAY151, which is biologically inactive. Only 33% of the drug is excreted by the kidneys, and the main route of elimination involves metabolism. Therefore, CKD may influence the pharmacokinetics of this drug [113, 114].
In subjects with varying degrees of renal impairment, systemic exposure to vildagliptin is increased approximately twofold compared with that in subjects with normal renal function; however, no correlation was observed between increase in exposure and severity of renal impairment. The lack of an obvious correlation may be a consequence of the kidneys’ contributions to both the excretion and the hydrolytic metabolism of vildagliptin [113, 114]. From a pharmacokinetic point of view, this approximately twofold increase in exposure suggests that the dose of vildagliptin for T2DM patients with moderate and severe renal insufficiency should be reduced to half of the daily dose for patients with normal renal function (50 mg once daily instead of 50 mg twice daily) [110, 115].
Alogliptin is a DPP-4 inhibitor that is approved in Japan for the treatment of adult patients with T2DM. The recommended dose of alogliptin is 25 mg once daily. Alogliptin is rapidly absorbed and unaffected by food, has an approximate bioavailability of 100%, and achieves its peak plasma concentration 1–2 h after administration. Alogliptin undergoes limited metabolism that involves the hepatic enzymes CYP2D6 and CYP3A4. Urinary excretion of the unchanged alogliptin is equal to 70% of the absorbed dose. Therefore, T2DM patients with renal impairment may require an appropriate dose reduction of vildagliptin. For patients with moderate renal impairment, the recommended dose is 12.5 mg once daily. In patients with severe renal impairment or ESRD, the recommended dose is 6.25 mg once daily [116, 117].
Linagliptin is a DPP-4 inhibitor that was approved by the US FDA in May 2011. It is administered at a therapeutic dose of 5 mg once daily. Linagliptin has a long half-life. It is metabolized into several inactive metabolites. Almost 85% of the dose undergoes fecal excretion via the enterohepatic system, whereas renal excretion is only a minor elimination pathway of linagliptin at therapeutic dose levels. Therefore, no dose adjustments are required in T2DM patients with impaired renal function, and linagliptin appears to be safe in T2DM patients with renal failure [118, 119].
The main pharmacokinetic parameters of DPP-4 inhibitors in T2DM subjects with renal impairment are summarized in Table 2, whereas dose adjustments in T2DM patients with renal impairment for all currently available DPP-4 inhibitors are summarized in Table 3.
7 Conclusion
Due to the increasing number of individuals with diabetes and diabetic complications, a high demand for novel and effective therapeutic strategies with the primary aim of delaying and/or ameliorating diabetic complications including renal impairment has emerged. All patients with diabetes and impaired renal function require dose adjustment of the administered antidiabetic drug. The exception is a DPP-4 inhibitor, linagliptin, which does not require dose optimization and is thus the most popular drug for diabetic patients with renal failure. Despite the results of numerous studies describing renoprotective effects of DPP-4 inhibitors in animal models of diabetic and nondiabetic nephropathy, there is still insufficient clinical evidence to confirm renal benefits. Large, randomized, controlled studies are needed to elucidate the effects of DPP-4 inhibitors on progressive kidney diseases, especially considering that DPP-4 inhibitors represent a chemically heterogeneous class of drugs that show significant pharmacological differences. In addition, the potential combined use of DPP-4 inhibitors with SGLT-2 inhibitors or RAAS modulators should be investigated further in models of renal impairment.
References
American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37(Suppl 1):S81–90.
Animaw W, Seyoum Y. Increasing prevalence of diabetes mellitus in a developing country and its related factors. PLoS One. 2017;12(11):e0187670-e. https://doi.org/10.1371/journal.pone.0187670.
Chawla A, Chawla R, Jaggi S. Microvasular and macrovascular complications in diabetes mellitus: distinct or continuum? Indian J Endocrinol Metab. 2016;20(4):546–51.
Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29(3):116–22.
Cade WT. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys Ther. 2008;88(11):1322–35.
Koye DN, Shaw JE, Reid CM, Atkins RC, Reutens AT, Magliano DJ. Incidence of chronic kidney disease among people with diabetes: a systematic review of observational studies. Diabet Med. 2017;34(7):887–901.
Ashby K, Navarro Almario EE, Tong W, Borlak J, Mehta R, Chen M. Review article: therapeutic bile acids and the risks for hepatotoxicity. Aliment Pharmacol Ther. 2018;47(12):1623–38.
Ritz E, Rychlik I, Locatelli F, Halimi S. End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am J Kidney Dis. 1999;34(5):795–808.
Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560–72.
Dreisbach AW, Lertora JJL. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74.
Ibrahim S, Honig P, Huang SM, Gillespie W, Lesko LJ, Williams RL. Clinical pharmacology studies in patients with renal impairment: past experience and regulatory perspectives. J Clin Pharmacol. 2000;40(1):31–8.
Nolin T. Drug metabolism in kidney disease. In: Xie W, editor. Drug metabolism in diseases. Amsterdam: Elsevier; 2017. p. 91–113.
Munar MY, Singh H. Drug dosing adjustments in patients with chronic kidney disease. Am Fam Physician. 2007;75(10):1487–96.
Lam YW, Banerji S, Hatfield C, Talbert RL. Principles of drug administration in renal insufficiency. Clin Pharmacokinet. 1997;32(1):30–57.
Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab. 1964;24:1076–82.
Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic sujbjects. J Clin Invest. 1967;46(12):1954–62.
Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63(2):492–8.
Williams JA. GLP-1 mimetic drugs and the risk of exocrine pancreatic disease: cell and animal studies. Pancreatology. 2016;16(1):2–7.
Holst JJ. Glucagon and glucagon-like peptides 1 and 2. Results Probl Cell Differ. 2010;50:121–35.
Brubaker PL. Glucagon-like peptide-2 and the regulation of intestinal growth and function. Compr Physiol. 2018;8(3):1185–210.
Paternoster S, Falasca M. Dissecting the physiology and pathophysiology of glucagon-like peptide-1. Front Endocrinol (Lausanne). 2018;9:584. https://doi.org/10.3389/fendo.2018.00584.
Karhus ML, Bronden A, Sonne DP, Vilsboll T, Knop FK. Evidence connecting old, new and neglected glucose-lowering drugs to bile acid-induced GLP-1 secretion: a review. Diabetes Obes Metab. 2017;19(9):1214–22.
Đanić M, Stanimirov B, Pavlovic N, Golocorbin-Kon S, Al-Salami H, Stankov K, et al. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol. 2018;9:1382. https://doi.org/10.3389/fphar.2018.01382.
Orskov C, Wettergren A, Holst JJ. Secretion of the incretin hormones glucagon-like peptide-1 and gastric inhibitory polypeptide correlates with insulin secretion in normal man throughout the day. Scand J Gastroenterol. 1996;31(7):665–70.
Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D, et al. Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes. 2004;53(3):654–62.
Deacon CF. Peptide degradation and the role of DPP-4 inhibitors in the treatment of type 2 diabetes. Peptides. 2018;100:150–7.
Tuch BE. Clinical use of GLP-1 agonists and DPP4 inhibitors. Pancreatology. 2016;16(1):8–9.
Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143(11):4397–408.
Tortosa F, Dotta F. Incretin hormones and beta-cell mass expansion: what we know and what is missing? Arch Physiol Biochem. 2013;119(4):161–9.
Dalsgaard NB, Vilsboll T, Knop FK. Effects of glucagon-like peptide-1 receptor agonists on cardiovascular risk factors: a narrative review of head-to-head comparisons. Diabetes Obes Metab. 2018;20(3):508–19.
Liu L, Shao Z, Xia Y, Qin J, Xiao Y, Zhou Z, et al. Incretin-based therapies for patients with type 1 diabetes: a meta-analysis. Endocr Connect. 2019;8(3):277–88. https://doi.org/10.1530/ec-18-0546.
Doupis J, Veves A. DPP4 inhibitors: a new approach in diabetes treatment. Adv Ther. 2008;25(7):627–43.
Deacon CF. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front Endocrinol (Lausanne). 2019;10:80. https://doi.org/10.3389/fendo.2019.00080.
Panchapakesan U, Pollock C. The role of dipeptidyl peptidase-4 inhibitors in diabetic kidney disease. Front Immunol. 2015;6:443. https://doi.org/10.3389/fimmu.2015.00443.
Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014;35(6):992–1019.
Mentlein R. Dipeptidyl-peptidase IV (CD26)—role in the inactivation of regulatory peptides. Regul Pept. 1999;85(1):9–24.
Ryan GJ, Moniri NH, Smiley DD. Clinical effects of once-weekly exenatide for the treatment of type 2 diabetes mellitus. Am J Health Syst Pharm. 2013;70(13):1123–31.
Sisson EM. Liraglutide: clinical pharmacology and considerations for therapy. Pharmacotherapy. 2011;31(9):896–911.
Romera I, Cebrian-Cuenca A, Alvarez-Guisasola F, Gomez-Peralta F, Reviriego J. A review of practical issues on the use of glucagon-like peptide-1 receptor agonists for the management of type 2 diabetes. Diabetes Ther. 2019;10(1):5–19.
Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–42.
Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60(4):470–512.
Scheen AJ. DPP-4 inhibitors in the management of type 2 diabetes: a critical review of head-to-head trials. Diabetes Metab. 2012;38(2):89–101.
Rohrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol. 2015;6:386. https://doi.org/10.3389/fimmu.2015.00386.
Gallwitz B. Sitagliptin: profile of a novel DPP-4 inhibitor for the treatment of type 2 diabetes. D Drugs Today (Barc). 2007;43(1):13–25.
Nabeno M, Akahoshi F, Kishida H, Miyaguchi I, Tanaka Y, Ishii S, et al. A comparative study of the binding modes of recently launched dipeptidyl peptidase IV inhibitors in the active site. Biochem Biophys Res Commun. 2013;434(2):191–6.
Berger JP, SinhaRoy R, Pocai A, Kelly TM, Scapin G, Gao YD, et al. A comparative study of the binding properties, dipeptidyl peptidase-4 (DPP-4) inhibitory activity and glucose-lowering efficacy of the DPP-4 inhibitors alogliptin, linagliptin, saxagliptin, sitagliptin and vildagliptin in mice. Endocrinol Diabetes Metab. 2018;1(1):e00002. https://doi.org/10.1002/edm2.2.
Makdissi A, Ghanim H, Vora M, Green K, Abuaysheh S, Chaudhuri A, et al. Sitagliptin exerts an antinflammatory action. J Clin Endocrinol Metab. 2012;97(9):3333–41.
Ommen ES, Xu L, O’Neill EA, Goldstein BJ, Kaufman KD, Engel SS. Comparison of treatment with sitagliptin or sulfonylurea in patients with type 2 diabetes mellitus and mild renal impairment: a post hoc analysis of clinical trials. Diabetes Ther. 2015;6(1):29–40.
Ueda S, Shimabukuro M, Arasaki O, Node K, Nomiyama T, Morimoto T. Effect of anagliptin and sitagliptin on low-density lipoprotein cholesterol in type 2 diabetic patients with dyslipidemia and cardiovascular risk: rationale and study design of the REASON trial. Cardiovasc Drugs Ther. 2018;32(1):73–80.
Kitada M, Tsuda SI, Konishi K, Takeda-Watanabe A, Fujii M, Kanasaki K, et al. Anagliptin ameliorates albuminuria and urinary liver-type fatty acid-binding protein excretion in patients with type 2 diabetes with nephropathy in a glucose-lowering-independent manner. BMJ Open Diabetes Res Care. 2017;5(1):e000391. https://doi.org/10.1136/bmjdrc-2017-000391.
Shinjo T, Nakatsu Y, Iwashita M, Sano T, Sakoda H, Ishihara H, et al. DPP-IV inhibitor anagliptin exerts anti-inflammatory effects on macrophages, adipocytes, and mouse livers by suppressing NF-kappaB activation. Am J Physiol Endocrinol Metab. 2015;309(3):E214–23.
McHugh KR, DeVore AD, Mentz RJ, Edmonston D, Green JB, Hernandez AF. The emerging role of novel antihyperglycemic agents in the treatment of heart failure and diabetes: a focus on cardiorenal outcomes. Clin Cardiol. 2018;41(9):1259–67.
Muskiet MHA, Tonneijck L, Smits MM, van Baar MJB, Kramer MHH, Hoorn EJ, et al. GLP-1 and the kidney: from physiology to pharmacology and outcomes in diabetes. Nat Rev Nephrol. 2017;13(10):605–28.
Tsimihodimos V, Karanatsis N, Tzavela E, Elisaf M. Antidiabetic drugs and the kidney. Curr Pharm Des. 2017;23(41):6310–20.
Muskiet MH, Smits MM, Morsink LM, Diamant M. The gut-renal axis: do incretin-based agents confer renoprotection in diabetes? Nat Rev Nephrol. 2014;10(2):88–103.
Hoorn EJ, Zietse R. Gut-kidney kaliuretic signaling: looking forward to feeding. Kidney Int. 2015;88(6):1230–2.
Michell AR, Debnam ES, Unwin RJ. Regulation of renal function by the gastrointestinal tract: potential role of gut-derived peptides and hormones. Annu Rev Physiol. 2008;70:379–403.
Yang J, Jose PA, Zeng C. Gastrointestinal-renal axis: role in the regulation of blood pressure. J Am Heart Assoc. 2017;6(3):e005536. https://doi.org/10.1161/JAHA.117.005536.
Jensen EP, Poulsen SS, Kissow H, Holstein-Rathlou NH, Deacon CF, Jensen BL, et al. Activation of GLP-1 receptors on vascular smooth muscle cells reduces the autoregulatory response in afferent arterioles and increases renal blood flow. Am J Physiol Renal Physiol. 2015;308(8):F867–77.
Schlatter P, Beglinger C, Drewe J, Gutmann H. Glucagon-like peptide 1 receptor expression in primary porcine proximal tubular cells. Regul Pept. 2007;141(1–3):120–8.
Gutzwiller JP, Tschopp S, Bock A, Zehnder CE, Huber AR, Kreyenbuehl M, et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J Clin Endocrinol Metab. 2004;89(6):3055–61.
Tonneijck L, Smits MM, Muskiet MHA, Hoekstra T, Kramer MHH, Danser AHJ, et al. Acute renal effects of the GLP-1 receptor agonist exenatide in overweight type 2 diabetes patients: a randomised, double-blind, placebo-controlled trial. Diabetologia. 2016;59(7):1412–21.
Nistala R, Savin V. Diabetes, hypertension, and chronic kidney disease progression: role of DPP4. Am J Physiol Renal Physiol. 2017;312(4):F661–70.
Girardi AC, Knauf F, Demuth HU, Aronson PS. Role of dipeptidyl peptidase IV in regulating activity of Na+/H+ exchanger isoform. N Am J Physiol Cell Physiol. 2004;287(5):C1238–45.
Marina AS, Kutina AV, Shakhmatoba EI, Natochin YV. Involvement of glucagon-like peptide-1 in the regulation of selective excretion of sodium or chloride ions by the kidneys. Bull Exp Biol Med. 2017;162(4):436–40.
Marina AS, Kutina AV, Shakhmatova EI, Balbotkina EV, Natochin YV. Stimulation of glucagon-like peptide-1 secretion by water loading in human. Dokl Biol Sci. 2014;459:323–5.
McKay NJ, Galante DL, Daniels D. Endogenous glucagon-like peptide-1 reduces drinking behavior and is differentially engaged by water and food intakes in rats. J Neurosci. 2014;34(49):16417–23.
Gutzwiller JP, Hruz P, Huber AR, Hamel C, Zehnder C, Drewe J, et al. Glucagon-like peptide-1 is involved in sodium and water homeostasis in humans. Digestion. 2006;73(2–3):142–50.
McKay NJ, Daniels D. Glucagon-like peptide-1 receptor agonist administration suppresses both water and saline intake in rats. J Neuroendocrinol. 2013;25(10):929–38.
Bankir L, Roussel R, Bouby N. Protein- and diabetes-induced glomerular hyperfiltration: role of glucagon, vasopressin, and urea. Am J Physiol Renal Physiol. 2015;309(1):F2–23.
Makino Y, Fujita Y, Haneda M. Dipeptidyl peptidase-4 inhibitors in progressive kidney disease. Curr Opin Nephrol Hypertens. 2015;24(1):67–73.
Tang-Christensen M, Larsen PJ, Göke R, Fink-Jensen A, Jessop DS, Møller M, et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol. 1996;271(440–4):R848–56.
Moreno C, Mistry M, Roman RJ. Renal effects of glucagon-like peptide in rats. Eur J Pharmacol. 2002;434(3):163–7.
du Cheyron D, Chalumeau C, Defontaine N, Klein C, Kellermann O, Paillard M, et al. Angiotensin II stimulates NHE3 activity by exocytic insertion of the transporter: role of PI 3-kinase. Kidney Int. 2003;64(3):939–49.
Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Cell Physiol. 2010;298(1):F177–86.
Girardi AC, Fukuda LE, Rossoni LV, Malnic G, Reboucas NA. Dipeptidyl peptidase IV inhibition downregulates Na+–H+ exchanger NHE3 in rat renal proximal tubule. Am J Physiol Cell Physiol. 2008;294(2):F414–22.
Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, Krause-Relle K, et al. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press Res. 2012;36(1):119–30.
Crajoinas RO, Polidoro JZ, Carneiro de Morais CP, Castelo-Branco RC, Girardi AC. Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am J Physiol Cell Physiol. 2016;311(5):C768–76.
Crajoinas RO, Oricchio FT, Pessoa TD, Pacheco BPM, Lessa LMA, Malnic G, et al. Mechanisms mediating the diuretic and natriuretic actions of the incretin hormone glucagon-like peptide-1. Am J Physiol Renal Physiol. 2011;301(2):F355–63.
Farah LXS, Valentini V, Pessoa TD, Malnic G, McDonough AA, Girardi ACC. The physiological role of glucagon-like peptide-1 in the regulation of renal function. Am J Physiol Renal Physiol. 2015;310(2):F123F7.
Mima A, Hiraoka-Yamomoto J, Li Q, Kitada M, Li C, Geraldes P, et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCβ activation in diabetes. Diabetes. 2012;61(11):2967–79.
Rieg T, Gerasimova M, Murray F, Masuda T, Tang T, Rose M, et al. Natriuretic effect by exendin-4, but not the DPP-4 inhibitor alogliptin, is mediated via the GLP-1 receptor and preserved in obese type 2 diabetic mice. Am J Physiol Renal Physiol. 2012;303(7):F963–71.
Takashima S, Fujita H, Fujishima H, Shimizu T, Sato T, Morii T, et al. Stromal cell–derived factor-1 is upregulated by dipeptidyl peptidase-4 inhibition and has protective roles in progressive diabetic nephropathy. Kidney Int. 2016;90(4):783–96.
Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci. 2007;81(4):272–9.
Skovso S. Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J Diabetes Investig. 2014;5(4):349–58.
Kanasaki K. The role of renal dipeptidyl peptidase-4 in kidney disease: renal effects of dipeptidyl peptidase-4 inhibitors with a focus on linagliptin. Clin Sci. 2018;132(4):489–507.
Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP-4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos. 2009;30(5):229–40.
Gill A, Gray SP, Watson AMD, Cooper ME, Jandeleit-Dahm KAM. Renoprotective effects of linagliptin and empagliflozin in a rat model of early diabetic nephropathy. Diabetes. 2017;66:A130(492–P).
Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63(6):2120–31.
Mega C, Vala H, Rodrigues-Santos P, Oliveira J, Teixeira F, Fernandes R, et al. Sitagliptin prevents aggravation of endocrine and exocrine pancreatic damage in the Zucker Diabetic Fatty rat—focus on amelioration of metabolic profile and tissue cytoprotective properties. Diabetol Metab Syndr. 2014;6(1):42. https://doi.org/10.1186/1758-5996-6-42.
Liu WJ, Xie SH, Liu YN, Kim W, Jin HY, Park SK, et al. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J Pharmacol Exp Ther. 2012;340(2):248–55.
Zhang J, Chen Q, Zhong J, Liu C, Zheng B, Gong Q. DPP-4 inhibitors as potential candidates for antihypertensive therapy: improving vascular inflammation and assisting the action of traditional antihypertensive drugs. Front Immunol. 2019;10:1050. https://doi.org/10.3389/fimmu.2019.01050.
Tomovic K, Lazarevic J, Kocic G, Deljanin-Ilic M, Anderluh M, Smelcerovic A. Mechanisms and pathways of anti-inflammatory activity of DPP-4 inhibitors in cardiovascular and renal protection. Med Res Rev. 2019;39(1):404–22.
Sun F, Wu S, Wang J, Guo S, Chai S, Yang Z, et al. Effect of glucagon-like peptide-1 receptor agonists on lipid profiles among type 2 diabetes: a systematic review and network meta-analysis. Clin Ther. 2015;37(1):225–41.
Monami M, Lamanna C, Desideri CM, Mannucci E. DPP-4 inhibitors and lipids: systematic review and meta-analysis. Adv Ther. 2012;29(1):14–25.
Liu SC, Tu YK, Chien MN, Chien KL. Effect of antidiabetic agents added to metformin on glycaemic control, hypoglycaemia and weight change in patients with type 2 diabetes: a network meta-analysis. Diabetes Obes Metab. 2012;14(9):810–20.
Aaboe K, Knop FK, Vilsbøll T, Deacon CF, Holst JJ, Madsbad S, et al. Twelve weeks treatment with the DPP-4 inhibitor, sitagliptin, prevents degradation of peptide YY and improves glucose and non-glucose induced insulin secretion in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2010;12(4):323–33.
Zhang X, Zhao Q. Effects of dipeptidyl peptidase-4 inhibitors on blood pressure in patients with type 2 diabetes: a systematic review and meta-analysis. J Hypertens. 2016;34(2):167–75.
McGuire DK, Alexander JH, Johansen OE, Perkovic V, Rosenstock J, Cooper ME, et al. Linagliptin effects on heart failure and related outcomes in individuals with type 2 diabetes mellitus at high cardiovascular and renal risk in CARMELINA. Circulation. 2019;139(3):351–61.
Rosenstock J, Perkovic V, Johansen OE, Cooper ME, Kahn SE, Marx N, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321(1):69–79.
Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, et al. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation. 2014;130(18):1579–88.
Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385(9982):2067–76.
McGuire DK, Van De Werf F, Armstrong PW, Standl E, Koglin J, Green JB, et al. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1(2):126–35.
Bergman A, Ebel D, Liu F, Stone J, Wang A, Zeng W, et al. Absolute bioavailability of sitagliptin, an oral dipeptidyl peptidase-4 inhibitor, in healthy volunteers. Biopharm Drug Dispos. 2007;28(6):315–22.
Pathak R, Bridgeman MB. Dipeptidyl peptidase-4 (DPP-4) inhibitors in the management of diabetes. P T. 2010;35(9):509–13.
Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP. Sitagliptin, a DPP-4 inhibitor for the treatment of patients with type 2 diabetes: a review of recent clinical trials. Curr Med Res Opin. 2008;24(2):489–96.
Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther. 2005;78(6):675–88.
Bergman AJ, Cote J, Yi B, Marbury T, Swan SK, Smith W, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care. 2007;30(7):1862–4.
Chan JCN, Scott R, Arjona Ferreira JC, Sheng D, Gonzalez E, Davies MJ, et al. Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab. 2008;10(7):545–55.
Gupta V, Kalra S. Choosing a gliptin. Indian J Endocrinol Metab. 2011;15(4):298–308.
Boulton DW. Clinical pharmacokinetics and pharmacodynamics of saxagliptin, a dipeptidyl peptidase-4 inhibitor. Clin Pharmacokinet. 2017;56(1):11–24.
Boulton B, Li L, Frevert EU, Tang A, Castaneda L, Vachharajani NN, et al. Influence of renal or hepatic impairment on the pharmacokinetics of saxagliptin. Clin Pharmacokinet. 2011;50(4):253–65.
He YL. Clinical pharmacokinetics and pharmacodynamics of vildagliptin. Clin Pharmacokinet. 2012;51(3):147–62.
He H, Tran P, Yin H, Smith H, Batard Y, Wang L, et al. Absorption, metabolism, and excretion of [14C]vildagliptin, a novel dipeptidyl peptidase 4 inhibitor, in humans. Drug Metab Dispos. 2009;37(3):536–44.
Russo E, Penno G, Del Prato S. Managing diabetic patients with moderate or severe renal impairment using DPP-4 inhibitors: focus on vildagliptin. Diabetes Metab Syndr Obes. 2013;6:161–70.
Dineen L, Law C, Scher R, Pyon E. Alogliptin (nesina) for adults with type-2 diabetes. P T. 2014;39(3):186–202.
Sakai Y, Suzuki A, Mugishima K, Sumi Y, Otsuka Y, Otsuka T, et al. Effects of alogliptin in chronic kidney disease patients with type 2 diabetes. Intern Med. 2014;53(3):195–203.
Barnett AH. Linagliptin: a novel dipeptidyl peptidase 4 inhibitor with a unique place in therapy. Adv Ther. 2011;28(6):447–59.
Blech S, Ludwig-Schwellinger E, Grafe-Mody EU, Withopf B, Wagner K. The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab Dispos. 2010;38(4):667–78.
He YL, Kulmatycki K, Zhang Y, Zhou W, Reynolds C, Ligueros-Saylan M, et al. Pharmacokinetics of vildagliptin in patients with varying degrees of renal impairment. Int J Clin Pharmacol Ther. 2013;51(9):693–703.
Naik H, Czerniak R, Vakilynejad M. Application of pharmacometric approaches to evaluate effect of weight and renal function on pharmacokinetics of alogliptin. Br J Clin Pharmacol. 2016;81(4):700–12.
Graefe-Mody U, Friedrich C, Port A, Ring A, Retlich S, Heise T, et al. Effect of renal impairment on the pharmacokinetics of the dipeptidyl peptidase-4 inhibitor linagliptin. Diabetes Obes Metab. 2011;13(10):939–46.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was supported by HORIZON 2020 MEDLEM project no. 690876, Project for Scientific and Technological Development of Vojvodina no. 114-451-2072-/2016-02, and Project of Ministry of Education, Science and Technological Development of the Republic of Serbia, Grant no. III41012.
Conflict of Interest
The authors of the manuscript (MM, NP, BS, MĐ, SGK, KS, and HAS) have no commercial associations or other arrangements that may pose a conflict of interest in connection with the article.
Rights and permissions
About this article

Cite this article
Mikov, M., Pavlović, N., Stanimirov, B. et al. DPP-4 Inhibitors: Renoprotective Potential and Pharmacokinetics in Type 2 Diabetes Mellitus Patients with Renal Impairment. Eur J Drug Metab Pharmacokinet 45, 1–14 (2020). https://doi.org/10.1007/s13318-019-00570-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-019-00570-y