
Abstract
Eperisone hydrochloride (4′-ethyl-2-methyl-3-piperidinopropiophenone hydrochloride) is a muscle relaxant agent, widely used in the treatment of patients with muscular contractures, low back pain or spasticity. Because of its mechanism of action (inhibition of gamma-efferent firing and local vasodilatation activity), side effects on central nervous system are rarely observed. A sensitive liquid chromatography–electrospray ionization-mass spectrometry method for determination of eperisone in human plasma has been developed, with a lower limit of quantification of 0.01 ng/mL. The method was applied to a pharmacokinetic study in 12 healthy volunteers given eperisone 100 mg as single dose on day 1 and three times daily on days 2 to 4. Eperisone was rapidly absorbed after oral administration (T max = 1.6 h) as it was expected by its fast-onset relaxant activity. Moreover, eperisone underwent a rapid elimination from the body (biological half-life 1.87 h), which was not modified during the repeated dosing as suggested by the C max cumulation observed, not different from that expected for a t 1/2 of 1.87 h as suggested by the similar and negligible plasma concentration values (0.063 and 0.067 ng/mL) measured on day 4 before the morning dose and 12 h after evening dose, thus ruling out any potential risk for drug accumulation. Thus, the pharmacokinetic characteristics of eperisone provide further justification for its tolerability in patients with low back pain or spastic palsy, in which the drug is given for periods ranging from few days to several months, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Low back pain (LBP) is generally thought to result from a self-perpetuating cycle of pain and muscle spasm, which is triggered by a pathological lesion in the lumbosacral region (e.g. facet joints, discs, muscles, or ligaments). The spastic palsy is also a clinical condition characterized by a velocity-dependent increase of muscle tone due to “parapyramidal” disturbance of the inhibitory afferents to the second motor neuron.
In both conditions, central muscle relaxant agents are widely used by physicians, although their use has been questioned by the occurrence of clinically relevant side effects mostly affecting the central nervous system (CNS), such as sedation, drowsiness, headache, blurred vision, nausea, and vomiting. Potential for drug abuse and dependency have been also reported (Elder 1991). In spite of this concern, however, many clinical guidelines still recommend the use of muscle relaxants, either alone or in combination with anti-inflammatory drugs for the management of LBP, and numerous physicians report the use of muscle relaxants although they are conditionally discouraged by their well known side effects (Di Iorio et al. 2000).
Eperisone hydrochloride (4’-ethyl-2-methyl-3-piperidinopropiophenone hydrochloride) is used clinically in the treatment of patients with LBP (Beltrame et al. 2008; Cabitza and Randelli 2008; Sartini 2008; Viladot Pericé et al. 2008) or spastic palsy (Bresolin et al. 2009a, b). Its peculiar mechanism of action is believed to blockade both sodium and calcium voltage-gated channels (Kocsis et al. 2005), exerting its activity mainly on the spinal cord structures by reducing the gamma-efferent firing and inhibiting the spinal cord activities (Ito et al. 1985; Morikawa et al. 1987). Furthermore, eperisone (EPS) have also shown some vasodilator activity with a consequent increase in muscle blood perfusion (Iwase et al. 1992; Sakai et al. 2008), and it exerts an antinociceptive effect by antagonistic activity on P substance (Fujioka and Kuriyama 1985). Because of its mechanisms of action, EPS is almost devoid of the side effects on CNS that are often reported with other centrally acting muscle relaxants.
Published pharmacokinetic (PK) investigations have shown that oral EPS is rapidly absorbed from the gastrointestinal (GI) tract with time to peak plasma concentrations (T max) of 1.1 h (Li et al. 2004), 1.28 h (Min Kyo et al. 2007) and 1.5 h (Takamatsu et al. 1992). This is a valuable characteristic of eperisone, since an essential request by general physicians for all muscle relaxants is their rapid onset of action, especially in LBP patients.
In contrast, the peak plasma concentrations (C max) have been reported to range within a wider interval, from 1.25 (Min Kyo et al. 2007) to 3.08 ng/mL (Li et al. 2004), after oral administration of a single 100 mg tablet of eperisone. The discrepancies observed in the C max of EPS may result from the different assay methods employed in the PK studies, which have shown variable lower limits of quantification (LLOQ) ranging between 0.2 and 1 ng/mL in the most recent studies (Li et al. 2004; Min Kyo et al. 2007). These LLOQ are not sensitive enough for PK research because the C max of EPS in some patients can be only approximately 0.5 ng/mL, and often the plasma concentrations observed in the terminal elimination phase are below 0.2 ng/mL.
We have therefore developed a liquid chromatography–mass spectrometry triple quadrupole mass spectrometry (LC–MS/MS) assay method for a rapid and sensitive determination of EPS, with the target LLOQ of 0.01 ng/mL in human plasma. Our aim was also to reduce the amount of human plasma and the time of sample processing needed for the assay of EPS. We have also taken the opportunity of this sensitive assay method to further investigate the PK of EPS when administered as single dose or as repeated dose three times a day after meals according to the regimen suggested for therapy; in particular, we wanted to gather more information on its potential for drug accumulation.
2 Materials and methods
2.1 Subjects
This study was carried out on 12 male healthy non-smoking volunteers aged 18–50 years. Subjects were called to participate in the study after detailed appropriate information on the nature and characteristics of the drug, as well as on the intention and executive formalities of the trial. The study was conducted at the University of Catania Medical Center (UCMC), in accordance with the protocol, GCP requirements and the principles of the Declaration of Helsinki, and the applicable regulatory requirements. The study protocol was reviewed and approved by the UCMC Ethical Committee. Informed written consent was obtained from all volunteers. The aim of the investigation was fully explained to all the subjects and informed consent was obtained in each case. Before treatments, and at the end of the study, all study subjects undertook a complete physical examination including systolic and diastolic blood pressure, heart rate (in sitting condition), electrocardiogram (ECG) and routine clinical laboratory analyses. The laboratory analyses included haematology, blood chemistry (urea, glucose, creatinine, bilirubin total, glutamic-oxalacetic transaminase, glutamic-pyruvic transaminase, cholesterol, and triglycerides), virology (HBV, HCV and HIV), urine analysis and screening for drugs abuse (methadone, benzodiazepines, cocaine, amphetamine/methamphetamine, opiates, barbiturates, tetrahydrocannabinol, and tricyclic antidepressants).
2.2 Study design
Aim of the study was to assess the pharmacokinetic profile of EPS after single (on day 1) and repeated (t.i.d. on days 2 to 4) oral administration of eperisone hydrochloride 100 mg tablets. Since the t 1/2 reported in the SPC and obtained in the present study was less than 2 h, the wash out period applied between the two phases of the study was considered adequate.
Twelve male healthy subjects, 18–50 years old, were enrolled. Each subject was given 100 mg tablets of eperisone hydrochloride after meal, in a single dose on day 1 and at the dose of three tablets daily for the next 3 days. In detail, EPS was administered at 8.00 a.m. on day 1, and at 8.00 a.m., 2.00 p.m. and 8.00 p.m. on days 2 to 4.
Blood samples were collected (Fig. 1):

Flow-chart of the PK study
-
On day 1: before administration (pre-dose) and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, and 12 h after dosing;
-
on day 4: before the 1st dosing at time 0 (pre-dose), and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6 h after dosing and before the 3rd dosing at time 0 (pre-dose) and at 0.5, 1, 1.5, 2, 2.5, 3, 12 h after dosing.
Each blood sample was immediately centrifuged at 4,100 rpm for 5 min and each plasma sample was separated and stored at −20° C until analyzed.
2.3 Analytical method
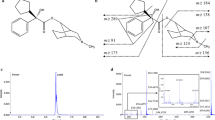
EPS plasma concentrations were determined using a LC–MS/MS equipped (Agilent 1100 series HPLC and an Agilent 6410 series Triple Quad.) with an electrospray ionization (ESI) source (Agilent Technologies, USA) and tolperisone (TOL) as internal standard (Min Kyo et al. 2007). The chromatographic separation was achieved on a Gemini-NX C18 (50 × 2.0 mm, 3 μm) column. All data were acquired employing Agilent 6410 Quantitative Analysis version B.01.03 analyst data processing software. The investigator who analyzed plasma samples was blind to treatment. EPS was extracted from plasma according to the following procedure. 500 μl of plasma in glass tubes were added with 10 μl (250 ng/mL) TOL, the mixture was vortex-mixed for approximately 1 min and added with 500 μl of saturated sodium bicarbonate solution, the plasma samples were extracted with 3.5 ml of ethyl acetate in single glass tubes. This was then mixed on a shaker for 10 min and centrifuged for 10 min at 4,100 rpm. A 3 ml portion of the organic phase was transferred to glass tubes and evaporated at 50°C for 60 min with a vacuum system. The residue was reconstituted with 100 μl of 10 mM ammonium acetate buffer solution pH 4.1:methanol (50:50, v/v) as mobile phase and an aliquot of 5 μl was injected into the HPLC column. EPS was ionized under positive ionization conditions. The product ion mass spectra of EPS and the TOL are shown in Fig. 2. The predominant peaks in the primary ESI spectra of EPS and TOL correspond to the [M–H]+ ions at m/z 260.3 and 246.3, respectively. Both product ions for EPS and TOL had a m/z of 98.2. Chromatograms were integrated and the calibration curves were plotted as the peak area of EPS corrected for variation in amounts of internal standard observed, versus EPS concentration.

The product ion mass spectra of eperisone (EPS) and tolperisone (TOL)
2.4 Pharmacokinetic parameters
The following PK parameters were determined using non-compartmental analysis:
maximum plasma concentration (C max) and time of maximum concentration (T max) were determined observationally, area under plasma concentration–time curve was calculated by numerical integration using the linear trapezoidal rule as follows:
Estimates of the terminal half-life t 1/2 were calculated using the following equation:
t 1/2 = 0.693/λ z , where λ z is the terminal elimination rate constant calculated by regression analysis on the natural log of plasma values versus time, between a time point where log linear λ z elimination phase begins and the time at which the last concentration above the LLOQ occurred.
λ z was calculated as the slope multiplied by (−1).
The area under concentration–time curve from time zero to infinity was calculated according to the following equation:
Plasma concentrations that were below the LLOQ were treated as zero.
2.5 Statistical analysis
A descriptive statistical analysis was carried out on the above mentioned parameters. Analysis of variance (ANOVA) was performed in order to verify the effect of repeated t.i.d. administration on the parameters AUC and C max relative to single dose after logarithmic transformation, using the “general linear model” procedure. The parameter T max was evaluated by a non-parametric procedure, the Wilcoxon matched-pair ranks test. A P value of 0.05 or less was considered as indicative of a significant difference.
3 Results
3.1 Method of assay
The validation results indicate that the assay has an adequate selectivity, specificity and sensitivity.
The assay was linear over a range of 0.01–10 ng/mL for EPS. The results for the intra- and inter-assay precision (calculated using over 3 analytical runs at 4 concentration levels, n = 6) ranged from 1.94 to 9.79% and from 1.86 to 10.04%, respectively. The inter-assay accuracy ranged from −1.41 to 4.18%. The coefficients of variation (CV) were less than 15% for both drugs. The lower limit of quantification was 0.010 ng/mL. Recoveries of the analysis were determined at two different EPS concentrations. The matrix effect for the measurement of EPS and TOL were not significant.
3.2 Pharmacokinetic analysis
The mean values of EPS plasma concentrations at each sampling time point after administration of 100 mg tablets of eperisone hydrochloride are outlined in Fig 3. The mean values of EPS log plasma concentrations versus time are outlined in Fig. 4. PK parameters represent mean values of the data of each subject analyzed separately. When a single dose of 100 mg tablet was given orally immediately after meal, the T max averaged 1.67 ± 0.49 (SD) h (range 1–2.5). The C max was 1.61 ng/mL ± 1.17 (range 0.40–3.77), the terminal elimination half-life (t ½) was 1.87 ± 0.73 h (range 0.95–3.53), and the AUC0–∞ was 2.79 ± 2.12 ng/ml/h (range 0.68–7.14) (Table 1). The drug plasma concentration measured immediately before the first dose of day 4 was on average 0.063 ± 0.048 ng/mL; this suggests that the drug accumulation during a t.i.d. administration is negligible. Moreover, the plasma concentrations 12 h after the third dose of day 4 averaged 0.067 ± 0.048 ng/mL, thus suggesting that a steady-state condition is achieved, as expected from the t ½ of eperisone.

Mean plasma concentration–time profile of eperisone after oral administration at 100 mg tablets to the human volunteers

Mean eperisone log plasma concentrations versus time
The comparison of the AUC0–6 after the first administration of day 4 versus the AUC0–∞ of the single dose administration (day 1) does not show any increase in drug exposure (2.674 vs. 2.798 ng/mL/h) (Table 1). In order to check the intra-day drug accumulation after a t.i.d. administration, a comparison of C max after the first and third dose has been performed on day 4. The mean C max values were 1.636 ± 1.299 and 1.715 ± 1.469 ng/mL, respectively, being the difference not statistically significant. The mean C max after the single dose of day 1 was 1.611 ± 1.166 ng/mL; also in this case there was no statistically significant difference between the C max values at day 1 and day 4 (Table 1). No serious adverse event was reported, and no deviation from normal values was observed in the lab exams. Only three subjects claimed of minor headache and epigastric pain that were considered possibly related to the drug administration.
4 Discussion
Eperisone hydrochloride, a central muscle relaxant, is widely used for the treatment of spasticity and muscle contracture and it is administered orally at a dose of 100 mg, usually at 300 mg/day (Beltrame et al. 2008; Cabitza and Randelli 2008; Sartini 2008; Viladot Pericé et al. 2008; Bresolin et al. 2009a, b). The aim of the study was to assess the pharmacokinetic profile of EPS at the dose of 100 mg tablet of eperisone hydrochloride administered in single and multiple doses.
A rapid and sensitive liquid chromatography–electrospray ionization-mass spectrometry method for the determination of EPS in human plasma was set up. The LLOQ was 0.01 ng/mL.
The results of the PK analysis in human volunteers confirm that EPS is rapidly absorbed after oral administration with meals; the T max occurs at about 100 min, suggesting that at least part of the drug absorption could have taken place in the stomach. A short T max after oral administration is an essential request by general physicians for all these compounds, especially in the treatment of LBP patients which ask for a fast-onset muscle relaxant and analgesic activity. For such a reason, parenteral formulations are usually preferred should the drug not guarantee a fast absorption from the GI tract after oral administration.
In our study, the T max value of EPS was comparable to those observed in other previous investigations (Li et al. 2004; Min Kyo et al. 2007; Takamatsu et al. 1992), and it was similar to the T max reported for other oral fast-acting muscle relaxants, such as tizanidine (1 h in fasting subjects and 2–3 h after a meal) (RxList, the Internet Drug Index 2010), baclofen (1.9 h) (Wuis et al. 1990) or thiocolchicoside (1 h) (Trellu et al. 2004). However, EPS seems to show some advantages over other muscle relaxant agents in terms of systemic tolerability. Since central muscle relaxants are used in repeated administration in LBP patients and chronically in the therapy of spastic palsy, the tolerability profile represents a central issue in the choice of the optimal treatment strategy.
EPS has been recently associated with a lower incidence of adverse events than thiocolchicoside, diazepam and baclofen (Cabitza and Randelli 2008; Viladot Pericé et al. 2008; Marchettini and Pecori 2009), confirming the good tolerability profile for eperisone, while over time many patients do not tolerate the side effects of other muscle relaxant agents (Tariq et al. 2005). Reasons for the better safety/risk ratio of EPS are probably related to its mechanism of action (inhibition of gamma-efferent firing on the spinal cord structure and local vasodilator activity with no CNS involvement) and, according to our observations, to its PK behavior as well. In fact, EPS is characterized by a biological half-life of 1.87 h in our study and similar values are reported in previous studies (Li et al. 2004; Min Kyo et al. 2007). Furthermore, it has been verified that by administration of EPS t.i.d. with meals no accumulation different from that expected for a drug with a half-life of less than 2 h was observed. All together, the PK characteristics of EPS can provide further justification, in addition to its mechanism of action, for its profile of tolerability in patients with LBP or spastic palsy, in which the drug is given for periods of time ranging from few days to several months, respectively.
Abbreviations
- LBP:
-
Low back pain
- CNS:
-
Central nervous system
- EPS:
-
Eperisone
- PK:
-
Pharmacokinetic
- GI:
-
Gastrointestinal
- T max :
-
Time of maximum concentration
- C max :
-
Maximum plasma concentration
- ng:
-
Nanogram
- mL:
-
Millilitre
- mg:
-
Milligram
- LLOQ:
-
Lower limits of quantification
- LC–MS/MS:
-
Liquid chromatography–mass spectrometry triple quadrupole mass spectrometry
- UCMC:
-
University of Catania Medical Center
- GCP:
-
Good clinical practice
- ECG:
-
Electrocardiogram
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- t ½ :
-
Plasma concentration half-life
- SPC:
-
Summary of product characteristics
- rpm:
-
Revolutions per minute
- min:
-
minute
- HPLC:
-
High performance liquid chromatography
- ESI:
-
Electrospray ionization
- TOL:
-
Tolperisone
- µl:
-
Microliter
- mM:
-
Millimole
- v/v:
-
Volume to volume
- m/z :
-
Mass to charge
- AUC0–t :
-
Area under the plasma concentration curve administration to last observed concentration at time t
- λ z :
-
Terminal elimination rate constant
- AUC0–∞ :
-
Area under the plasma concentration curve extrapolated to infinity time
- C last :
-
Last quantified concentration
- ANOVA:
-
Analysis of variance
- AUC:
-
Area under the plasma concentration curve
- CV:
-
Coefficient of variation
- SD:
-
Standard deviation
- h:
-
Hour
References
Beltrame A, Grangiè S, Guerra L (2008) Clinical experience with eperisone in the treatment of acute low back pain. Minerva Med 99:347–352
Bresolin N, Zucca C, Pecori A (2009a) Efficacy and tolerability of eperisone in patients with spastic palsy: a cross-over, placebo-controlled, dose-ranging trial. Eur Rev Med Pharmacol Sci 13:365–370
Bresolin N, Zucca C, Pecori A (2009b) Efficacy and tolerability of eperisone and baclofen in spastic palsy: a double-blind randomized trial. Adv Ther 26:1–6
Cabitza P, Randelli P (2008) Efficacy and safety of eperisone in patients with low back pain: a double blind randomized study. Eur Rev Med Pharmacol Sci 12:229–235
Di Iorio D, Henley E, Doughty A (2000) A survey of primary care physician practice patterns and adherence to acute low back problem guidelines. Arch Fam Med 9(10):1015–1021
Elder NC (1991) Abuse of skeletal muscle relaxants. Am Fam Physician 44:1223–1226
Fujioka M, Kuriyama H (1985) Eperisone, an antispastic agent, possesses vasodilating actions on the guinea-pig basilar artery. J Pharmacol Exp Ther 235:757–763
Ito T, Hori M, Furukaa K, Karasawa T, Kadokawa T (1985) Pharmacological studies of 1-(2, 3-dimethyl-4-mthoxyphenyl)-2-methyl-3-(1-pyrrolidinyl)-1-propanone hydrochloride (AD-2239), a centrally acting muscle relaxant. Arch Int Pharmacodyn 275:105–122
Iwase S, Mano T, Saito M, Ishida G (1992) Effect of a centrally-acting muscle relaxant, eperisone hydrochloride, on muscle sympathetic nerve activity in humans. Funct Neurol 7:459–470
Kocsis P, Farkas S, Fodor L, Bielik N, Than M, Kolok S, Gere A, Csejtei M, Tarnawaet I (2005) Tolperisone-type drugs inhibit spinal reflexes via blockade of voltage-gated sodium and calcium channels. J Pharmacol Exp Ther 315:1237–1246
Li D, Xin W, Shengqiang Z, Jianpin S, Yindi Z (2004) Rapid and sensitive liquid chromatography–electrospray ionization-mass spectrometry method for the determination of eperisone in human plasma: method and clinical applications. J Chromatogr Sci 42:254–258
Marchettini P, Pecori A (2009) Eperisone cloridrato versus baclofene nel trattamento a lungo termine della paralisi spastica: uno studio randomizzato in doppio cieco. Therapeutic Evidence Based Giugno 2(3). http://www.wkhealth.it/teb/default.asp. Accessed 6 July
Min Kyo J, Eun Sook J, Nam Hee K, Chang-Soo K, Youn-Bok C, Yong-Moon L, Su-Youn A, Hwang-Eui C, Yong Hwa L, Jin Tat H, Gong-Cheul M (2007) Determination of eperisone in human plasma by liquid chromatography–ESI-tandem mass spectrometry. Arch Pharm Res 30:1174–1178
Morikawa K, Oshita M, Yamazaki M, Ohara N, Kato H, Ito Y, Kontani H, Koshiura R (1987) Pharmacological studies of the new centrally acting muscle relaxant 4’-ethyl-2-methyl-3-pyrrolidinopropiophenone hydrochloride. Arzneim Forsch Drug Res 37:331–336
RxList, the Internet Drug Index (2010). Web address: http://www.rxlist.com/script/main/hp.asp. Accessed on January 8
Sakai Y, Matsuyama Y, Nakamura H et al (2008) The effect of muscle relaxant on the paraspinal muscle blood flow: a randomized controlled trial in patients with chronic low back pain. Spine 33:581–587
Sartini S (2008) Open experience with a new myorelaxant agent for low back pain. Adv Ther 25:1–9
Takamatsu T, Yamazaki K, Kayano M, Takenaka F, Hasui M, Okhawa T (1992) Determination of eperisone in human plasma by gas chromatography-mass spectrometry. J Chromatogr 584:261–266
Tariq M, Akhtar N, Alì M, Rao S, Badshah M, Irshad M (2005) Eperisone compared to physiotherapy on muscular tone of stroke patients: a prospective randomized open study. J Pak Med Assoc 55:202–204
Trellu M, Filali-Ansary A, Françon D, Adam R, Lluel P, Dubruc C, Thénot JP (2004) New metabolic and pharmacokinetic characteristics of thiocolchicoside and its active metabolite in healthy humans. Fundam Clin Pharmacol 18:493–501
Viladot Pericé R, Rodriguez Boronat E, Ferrer Bosch F, Rusinyol FC (2008) Myorelaxant effects of eperisone and diazepam in the treatment of acute spinal muscle contracture: a comparative study. J Anaesth Clin Pharmacol 24:285–290
Wuis EW, Dirks MJM, Vree TB, Van der Kleijn E (1990) Pharmacokinetics of baclofen in spastic patients receiving multiple oral doses. Pharm Weekbl (sci) 12:71–74
Acknowledgments
We thank Dr. Giacomo Spignoli for his kind help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Melilli, B., Piazza, C., Vitale, D.C. et al. Human pharmacokinetics of the muscle relaxant, eperisone hydrochloride by liquid chromatography–electrospray tandem mass spectrometry. Eur J Drug Metab Pharmacokinet 36, 71–78 (2011). https://doi.org/10.1007/s13318-011-0034-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-011-0034-0