
Abstract
A highly sensitive and rapid method for the analysis of pantoprazole in human plasma using liquid chromatography coupled to tandem electrospray ionization mass spectrometry was developed. The procedure involves a simple protein precipitation method with methyl alcohol and separation by RP-HPLC. Detection was performed by positive ion electrospray ionization in multiple reaction monitoring mode, monitoring the transitions m/z 384.1 → 200.0 and m/z 346.1 → 198.0, for quantification of pantoprazole and IS, respectively. The standard calibration curves showed good linearity within the range of 5–5,000 ng mL−1. The lower limit of quantitation (LLOQ) was about 5 ng mL−1. The extractive recovery of pantoprazole from the biological matrix was more than 77.58% and the matrix effect was complied with relevant provision. The intra-day accuracy of the drug containing serum samples was more than 92.19% with a precision of 0.79–5.36%. The inter-day accuracy was 85.49% or more, with a precision of 0.91–12.67%. Intra and inter-day accuracy of the assay at four concentrations were 97.9–98.2% with a precision of 4.2–13.9%. This method offered good precision and accuracy and was successfully applied to the pharmacokinetic and bioequivalence studies of 40 mg of enteric-coated pantoprazole in 20 healthy Chinese volunteers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Pantoprazole,5-(difluoromethoxy)-2-[(3,4-dimethoxy-2-pyridyl)methylsulfinyl]-1H-benzimi- dazole, is a selective and irreversible proton pump inhibitor (PPIs) (Filipe et al. 2008; Osmair et al. 2004). These drugs are now universally considered the treatment option for erosive esophagitis associated with gastroesophageal reflux disease (GERD), as well as for the treatment of pathological hypersecretion associated with Zollinger–Ellison syndrome. These drugs share similar properties: acid-activation step, covalent binding to the proton pump of the gastric parietal cell via the production of covalent disulphide bonds and relatively stable inhibition of H+/K+-ATPase. Pantoprazole, the third proton pump inhibitor (PPI), is characterized by its favorable pharmacokinetic properties, and its low potential to interact with concomitantly administered drugs and the cytochrome P450 enzyme system. Pantoprazole inhibits acid more powerfully than histamine H(2) receptor antagonists and omeprazole.
The extraction reported to detect pantoprazole was solid-phase extraction (Filipe et al. 2008) or liquid–liquid extraction (Osmair et al. 2004; Xu et al. 2008). However, these methods presented some disadvantages such as low sensitivity, being expensive and time consuming. Therefore, a simple, rapid extraction–precipitation of protein was developed. The aim of the present investigation was to develop a new, simple, rapid and sensitive liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) method for the estimation of pantoprazole in human plasma. The method was applied to a bioequivalence study.
2 Materials and methods
2.1 Materials
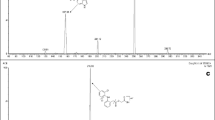
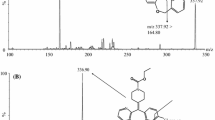
Pantoprazole (C16H14F2N3NaO4S MW 423.38 purity 94.6%) (Fig. 1a) and omeprazole (IS, C17H19N3O3S MW 345.41 purity 100.0%) (Fig. 1b) were purchased from the National Institute for The Control of Pharmaceutical and Biological Products (Beijing, China), respectively. The reference preparations (pantoprazole sodium enteric-coated capsules, 40 mg) were obtained from China and the United States East China Pharmaceutical Co., Ltd. (Hangzhou, East China) and the test preparations (pantoprazole sodium enteric-coated capsules, 40 mg) was provided by Kang Enbei Pharmaceutical Co., Ltd. (Hangzhou, East China). HPLC grade methanol was purchased from TEDIA. While all other reagents and solvents used were of analytical grade. Ultra pure water was obtained from Wahaha Group Co., Ltd. (Hangzhou, East China).

Structure of a pantoprazole and b internal standard omeprazole
2.2 Instruments
For analysis, an Agilent 1200SL Series liquid chromatographic system interfaced to an Agilent 6410B Triple Quad LC-ESI-MS/MS (MassHunter Data Acquisition, Qualitation and Quantitation soft-ware, USA) was quipped with an Lichrospher C18 column (5 μm, 2.1 × 100 mm) at a column temperature of 40°C. Other instruments used included WH-3 Micro Whirlpool mixer (Shanghai Huxi Analysis Instrument Factory Co. Ltd. China), Feige TGL-16C centrifuge (Shanghai Huxi Analysis Instrument Factory Co. Ltd. China).
2.3 Bioequivalence study design and procedures
The study was an open-randomized, balanced, two-period crossover experiment on healthy volunteers with a 1-week washout period. Twenty male healthy subjects with a mean age of 24.5 years (range of 18–27 years) and a mean weight of 64 kg (range of 55–74 kg) and a mean height of 172.15 cm (range of 160–184 cm) took part in this study after signing the consent form.
After an overnight fast, all subjects were given a single oral dose of 40 mg pantoprazole sodium enteric-coated test (Kang Enbei Pharmaceutical Co. Ltd.) or reference capsules (The United States East China Pharmaceutical Co. Ltd.) and continued fasting for 3 h after administration. A crossover study was followed by a washout period of 1 week. During the test period, all subjects remained under close medical supervision and were supplied uniform diets. Blood samples (3 mL) were taken before and at 0, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 6.0, 8.0, 10.0, 12.0 h after drug administration. The blood samples were collected in heparin-treated tubes, immediately subjected to centrifugation at 4,000 rpm min−1 for 15 min and then stored at −65°C until analysis.
2.4 Drug assay
2.4.1 Stock solutions and working solutions
A standard stock solution of pantoprazole was prepared by dissolving 20.01 mg pantoprazole sodium in 10 mL methanol to give a nominal concentration of 2 mg mL−1. The stock solution was appropriately diluted with methanol to obtain working solutions for calibration at 25, 50, 250, 500, 2,500, 5,000, 10,000 and 50,000 ng mL−1 of pantoprazole. The IS stock solution of omeprazole was prepared by dissolving about 20.0 mg omeprazole sodium in 10 mL methanol to give a nominal concentration of 2 mg mL−1. The IS working solution (about 800 ng mL−1) was prepared by diluting the stock solution of IS with methanol. The standard solutions of pantoprazole and omeprazole were protected from light and kept at 4°C before use.
2.4.2 Calibration standards and QC samples
Eight calibration standards ranging from 5 to 5,000 ng mL−1 were prepared by adding 10 μL of a known working solution of pantoprazole, according to “Sample preparation” The quality control samples were prepared in pool, at concentrations of 10 (low), 500 (medium) and 5,000 ng mL−1 (high), as a single batch at each concentration, and then divided in aliquots that were stored in the freezer at 4°C until analysis. They were used to check that the system performs correctly in control.
2.4.3 Sample preparation
All frozen human plasma samples were previously thawed at ambient temperature. A 50-μL volume of plasma was transferred to a microcentrifuge tube (1.5 mL), and then IS working solution and 200 mL methanol were sequentially added and vortex-mixed for 3 min. After centrifugation at 14,000 rpm min−1 for 10 min, the supernatant was transferred to vials on the rack of the autosampler and a 3 μL aliquot was injected into the column.
2.4.4 Chromatographic conditions
Mobile-phase composition was constituted of methanol:water (60:40, v/v), each containing 1‰ of 5 mol L−1 ammonium acetate) at a flow rate of 0.3 mL min−1. The column temperature was 40°C.
2.4.5 Mass-spectrometric conditions
The mass spectrometer was equipped with an electrospray source and set up in multiple reaction monitoring (MRM). The strongest multiple reaction monitoring transitions were selected with 384.1 → 200 and 346.1 → 198, for pantoprazole and omeprazole, respectively. In order to optimize all the MS parameters, a standard solution of the analyte and IS was infused into the mass spectrometer. For both pantoprazole and omeprazole, the following optimized parameters were obtained:
It was operated in positive ion mode using the following conditions: drying gas (N2) flow 6 L min−1, drying gas temperature 300°C, nebulizer pressure 30 psi, and ionspray voltage 4 kV. The fragmentor voltage used was 100 V. The collision energy was 6 and 8 eV for omeprazole and pantoprazole, respectively. Data acquisition and analysis were performed using Agilent Mass Hunter workstation (Mass Hunter Data Acquisition, Qualitation and Quantitation software, USA). Figure 2 shows the full scan positive ESI product ion mass spectra of the analytes.

Full-scan product ion spectra of: a pantoprazole and b omeprazole
2.5 Validation
2.5.1 Linearity
Linearity was evaluated using freshly prepared spiked plasma samples in the concentration range of 5–5,000 ng mL−1. Each calibration curve consisted of a drug-free human plasma sample at eight calibrator concentrations. Five such linearity curves were analyzed. In the plasma, pantoprazole standard curves were calculated by the equation fi = aC + b using weighted (1/response2) least square regression. A correlation of more than 0.99 was desirable for all the calibration curves.
2.5.2 Lower limit of quantitation
The lowest standard on the calibration curve was to be accepted as the lower limit of quantitation (LLOQ) if it complied the acceptance criteria of exhibiting the analyte response should be at least five times that of drug free (blank) processed plasma. LLOQ was defined as ten times the S/N (signal-to-noise ratio).
2.5.3 Specficity
Six randomly selected, control drug-free human plasma sample were processed by the similar extraction procedure and analyzed to determine the extent to which endogenous plasma components may contribute to the interference at retention time of analyte and IS.
2.5.4 Recovery and matrix effect
The recovery was evaluated by calculating the mean of the response of each concentration and dividing the extracted sample mean by the un-extracted (spiked blank plasma extract) sample mean of the corresponding concentration. Comparison with the un-extracted samples, spiked on plasma residues, was done in order to eliminate matrix effects, giving a true recovery. The matrix effect experiments were performed using the ratio between spiked mobile phase solutions and un-extracted samples spiked on plasma residues.
2.5.5 Accuracy and precision
Both the intra- and inter-day accuracy and precision evaluations were performed by repeated analysis of pantoprazole in human plasma. The run consisted of a calibration curve plus five replicates of each LLOQ, low-, medium- and high-quality control samples. The accuracy values for between- and within-batch studies were within acceptable limits (n = 5). The evaluation of precision was based on the criteria that the deviation of each concentration level should not be more than ±15.0% from the nominal concentration except for the LLOQ, for which it should not be more than ±20.0%. Similarly for accuracy, the mean value should not deviate by ±15.0% of the nominal concentration except for the LLOQ where it should not deviate by more than ±20.0% of the nominal concentration. The results shown in Table 1 indicate that the assay method is reproducible for replicate analysis of pantoprazole in hum plasma within the same day and reproducible on different days also (n = 5).
2.5.6 Stability
The stability of pantoprazole in plasma was evaluated in three studies: a short-term study, a long-term study and a freeze–thaw study. LLOQ and QC samples (10, 500, 5,000 ng mL−1) for pantoprazole were assayed five times. The concentration of pantoprazole after each storage period was related to the initial concentration as determined for samples that were freshly prepared and processed immediately. The stability of standard stock solution was also tested. The stock solutions were stable for at least 3 days when stored at 4°C. The stability experiments were aimed at testing all possible conditions that the samples might experience after collecting and prior the analysis.
2.5.7 Pharmacokinetic analysis
According to international guidelines, the main variables for testing the bioequivalence of two formulations are AUC and C max, calculated from the plasma concentrations of pantoprazole. T max has been considered a secondary variable. All the pharmacokinetic parameters were determined by non-compartmental analysis. Treatment sequence was tested against subjects within the treatment sequence to investigate any carryover effects. Point estimates and an associated 90% confidence interval were obtained in the log-scale for the difference ‘test minus reference drug’ using the residual variance. Point estimates and associated 90% confidence intervals obtained from the log-scale were then back-transformed to estimate the ‘test/reference’ ratios. Bioequivalence was accepted in case the 90% confidence intervals fell within the pre-defined limits of 80–125%. C max was assessed in a similar fashion. As T max is a discontinuous measure, confidence interval was calculated by a non-parametric approach and formulations were considered bioequivalent if the 90% confidence interval fell within the limits of 80–120%. Results throughout are expressed as mean ± standard deviation of mean (SD). The limit we have used (70–143%) is based on a local regulatory authority (SFDA 2005).
The BAPP 3.0 which was organized by the research center of China Pharmaceutical University Pharmacokinetics was used to calculate the PK. Non-compartmental PK analysis was employed to analyze plasma drug concentration–time data. The parameters C max and T max were obtained directly from experimental observations. The apparent elimination rate constant (λz) was obtained by the slope of the end of the straight part of logarithmic concentration–time curve. t 1/2, AUC and MRT were estimated by the following formulation:
C n is the final sampling point concentration.
3 Results and discussion
3.1 Drug assay
3.1.1 Linearity
The peak area ratio of pantoprazole to IS in human plasma was linear with respect to the analyte concentration over the range 5–5,000 ng mL−1. The mean correlation coefficient was 0.9960.
3.1.2 Lower limit of quantitation
The limit of quantitation (LOQ) was determined as the lowest concentration of pantoprazole in human plasma that could be quantified with acceptable precision and accuracy under the experimental conditions with a signal-to-noise ratio of at least 10:1. The lower limit of quantitation was about 5 ng mL−1 (lowest standard level).
3.1.3 Specificity
The analyte and IS were well separated under the described chromatographic conditions at retention time (RT) of 1.9 and 2.1 min, respectively. The total run time was 2.5 min. Figure 3 shows the representative chromatograms of blank plasma, standard solution of pantoprazole (50 ng mL−1) and IS (800 ng mL−1) dissolved in methanol, standard solution of pantoprazole (50 ng mL−1) and IS (800 ng mL−1) added in blank plasma and plasma sample obtained from a healthy subject after 3 h following an oral 40 mg dose of pantoprazole. The peaks were of good shape completely. No interference with constituents from the plasma matrix was observed.

Representative HPLC chromatogram of a blank plasma, b methanol spiked with pantoprazole and internal standard, c blank plasma spiked with pantoprazole and internal standard omeprazole, and d 3 h after administration (the internal standard omeprazole retention time is about 2.1 min, pantoprazole retention time is about 1.9 min)
3.1.4 Recovery and matrix effect
In LC-MS/MS we need to investigate the impact of matrix effects using appropriate methods to avoid or reduce this interference. Most of the previous reports treated samples using liquid–liquid extraction. We believed that compared to protein precipitation liquid–liquid extraction can be more thoroughly remove the interference matrix, but with operation complicated, time-consuming, reagent toxicity, high cost disadvantage. In pre-study, we used different ways to deal with plasma samples (liquid–liquid extraction with methylene chloride and tert-butyl ether, protein precipitation with methanol). Then treated samples were determined and compared. The result was that the methanol protein precipitation method has a smaller matrix effects and higher sensitivity. In order to further reduce the matrix effect, we took simple measures of reducing sample volume and injection volume. Only 50 μL of the plasma sample was precipitated with methanol, injecting 3 μL. No significant matrix effect was observed. The extraction recoveries pantoprazole in human plasma ranged from 78.63–109.7% (Table 1).
3.1.5 Accuracy and precision
The recovery for inter-day accuracy was between 85.49 and 117.94% with the relative standard deviation (RSD) of 2.81–4.37% in human plasma. Intra-day accuracy was between 82.91 and 86.69% with the RSD of 6.89–9.54% (Table 2).
3.2 Stability
3.2.1 Long-and short-term stability
Pantoprazole was stable at −65°C for 16 days in human plasma. The mean concentration of pantoprazole was 5.8440, 11.113, 511.86 and 4785.9 ng mL−1 at 16 days. When kept in the autosampler pantoprazole was found to be stable in 12.0 h in human plasma. The RSD was 6.44, 7.59, 1.34 and 1.55% at the concentrations of 5.2525, 10.505, 525.25 and 5252.5 ng mL−1, respectively.
3.2.2 Repeated freeze–thaw stability
Plasma samples of pantoprazole were found to be stable even after subjecting them to three freeze–thaw cycles. The mean concentration of pantoprazole was 5.4492, 10.731, 533.94 and 4662.0 ng mL−1 after three cycles. The percent RSD was 2.97, 7.07, 0.41 and 5.34% at the concentrations of 15.2525, 10.505, 525.25 and 5252.5 ng mL−1, respectively.
3.2.3 Stock solution stability
The stock solutions were stable for at least 3 days when stored at 4°C.
3.2.4 Statistical evaluation of pharmacokinetic parameters
Safety was evaluated by monitoring adverse events during the whole bioequivalence study in the clinic. The developed LC-MS/MS method had been successfully used in a pilot bioequivalence study of pantoprazole preparations in humans following oral administration.
Both the mean values and standard deviations of the main pharmacokinetic parameters such as C max, T max, AUC0−12 and AUC0−∞ were found to be close between test and reference preparations. Nearly all the bioequivalence parameters obtained in this study were similar to those reported in other literature using Chinese volunteers (Xu et al. 2008; Wang et al. 2006; Ling et al. 2005. In addition, the calculated 90% confidence interval for mean C max, AUClast and AUC0−∞ of two drugs lay within the FDA’s accepted range of 80–125%. Therefore, it could be concluded that the two pantoprazole preparations analyzed were bioequivalent in terms of rate and extent of absorption. Table 3 shows the bioequivalence parameters obtained. Figure 4 shows the mean plasma concentration versus time graphs of pantoprazole after administration of test and reference preparations to healthy, adult, male human volunteers under fasting condition.

Mean plasma concentration versus time graphs of pantoprazole after administration of test and reference preparations to healthy, adult, male and human volunteers under fasting condition
According to the applicable European guidelines, bioequivalence evaluation of two pantoprazole enteric-coated formulations should include a single-dose, 2-way crossover study under fed conditions (standardized high-fat meal) in addition to a fasting study (CPMP 1999). Similarly, the US Food and Drug Administration also requires a fed study for pantoprazole (FDA 2008), which is in line with the recommendations of the applicable guidance document (CDER 2002).
4 Discussion
The fact that the mobile phase contained a low amount of ammonium acetate did not interfere with the analysis. The presence of the acid was necessary in order to improve the detection of the compounds in positive electrospray.
The method takes full advantage of the benefits of LC-MS/MS as it enables the accurate, simple, rapid and selective assay of pantoprazole from the biological matrix. Only 50 μL volume of plasma was taken for sample preparation that greatly reduced the impact of matrix effects. Impurities in human plasma have no interference to analysis using a precipitation of protein with methanol. Comparing with other sample preparation methods reported, the one-step protein precipitation with methanol has overcome the shortcomings such as being of low sensitivity, time consuming and costly. In addition, this method permits an increase in sensitivity and specificity and can be carried out in a shorter time (RT of 1.9 min for pantoprazole and 2.1 min for omeprazole). No significant metabolic compounds were found to interfere with the analysis. Also, the LOQ observed (5 ng mL−1) is sufficient for bioequivalence studies. All samples’ plasma drug concentration was detected in bioequivalence study. Thus, this is a user-friendly method of analysis, particularly applicable to plasma concentration monitoring and bioavailability and pharmacokinetics study. The method was validated and it satisfied the requirement of linearity, recovery, accuracy, precision and stability for a bioequivalence study. The statistical analysis of pharmacokinetic parameters confirmed that the test preparations were bioequivalent with the reference in terms of rate and extent of absorption. The relative bioavailability of test preparations was 98.2 ± 18.4%.
There was paper reported of bioequivalence study of two enteric-coated formulations of pantoprazole in healthy volunteers under fed conditions (Filipe et al. 2008). Compared with the bioequivalence parameters obtained from this study, significant differences could be found in t max. When under fed conditions t max was much later. It was reported that when pantoprazole is given with food, its t max is highly variable and may increase significantly (Pantoprazole, Pub Drug http://www.smbrower.com/mediawiki/index.php/Pantoprazole). Therefore, it is worth mentioning that the food effects on the pharmacokinetics of pantoprazole reported elsewhere were shown in our results. In addition, differences also could be found in other bioequivalence parameters in those studies under two different conditions. Considering the volunteers were from Finland and China, we suspect that the racial difference was perhaps the main reason.
5 Conclusion
A sensitive and rapid LC-ESI-MS/MS method for pantoprazole quantitation in human plasma was validated. The method involved a sample preparation with adequate recovery by one-step protein precipitation with methanol and no significant matrix effect was observed. This method provided superior sensitivity with the lower limit of quantitation as low as 5 ng mL−1 for pantoprazole, and it had been successfully applied to pharmacokinetics and bioequivalence study.
References
Cente for Drug Evaluation and Research (CDER)-Food and Drug Administration (FDA) (2002) Guidance for industry: food-effect bioavailability and fed bioequivalence studies
Committee for Proprietary Medicinal Products (CPMP) (1999) Note for guidance on modified release oral and transdermic dosage forms: section II (pharmacokinetic and clinical evaluation). CPMP/EWP/280/96
Filipe A, Almeida S, Franco Spínola AC, Neves R, Trabelsi F, Torns A et al (2008) Bioequivalence study of two enteric-coated formulations of pantoprazole in healthy volunteers under fed. J Arzneimittel-Forschung (Drug Research) 58(9):451–456
Food and Drug Administration (FDA) (2008) Guidance on pantoprazole sodium, May 2008
Ling J, Wang H, Shentu JZH (2005) Determination of pantoprazole in human plasma by HPLC-MS.J. Chin J Mod Appl Pharm 22(4):306–308
Osmair P, Oliveira CH, Barrientos-Astigarraga RE, Rezende VM, Mendes GD et al (2004) Determination of pantoprazole in human plasma by LC-MS-MS using lansoprazole as internal standard. J Arzneimittel-Forschung (Drug Research) 54(6):314–319
SFDA guidance for industry. Bioavailability studies for orally administered drug-products (2005) General considerations. China Department of Health and Human Services, State Food and Drug administration, Centre for Drug Evaluation and Research 2005. http://www.sfda.gov.cn/WS01/CL0001/
Wang LX, Li JH, Li YB (2006) Bioequivalence of pantoprazole enteric capsules in human plasma. J Clin Med Pract 10(3):30–32
Xu HY, Yang L, Zhang P, Shen T, Zhao HQ (2008) Determination of pantoprazole in human plasma by HPLC.J. J Shenyang Pharm Univ 25(1):44–47
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Y., Ding, MJ., Ma, J. et al. Quantification of pantoprazole in human plasma using LC-MS/MS for pharmacokinetics and bioequivalence study. Eur J Drug Metab Pharmacokinet 35, 147–155 (2011). https://doi.org/10.1007/s13318-010-0018-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-010-0018-5