
Abstract
The soil-borne ascomycete Gaeumannomyces graminis var. tritici causes take-all of wheat (Triticum aestivum). Between host crops, G. graminis var. tritici survives saprophytically on crop debris and by infecting susceptible grass weeds or cereal volunteers. Invasion of roots in the following wheat crop results in reduced grain yield and quality. Take-all is commonly assessed in the field by visual inspection. Molecular-based methods are also available to detect G. graminis var. tritici, including a quantitative PCR (qPCR) assay that indirectly measures the amount of pathogen DNA in environmental samples. The qPCR is used as part of a commercial tool (known as PreDicta B™) to predict the risks of take-all in wheat crops prior to planting, which are dependent on the amount of Gaeumannomyces inoculum in field soils. Unfortunately, the costs associated with the PreDicta B™ test can be prohibitive to its use. As a result, in this study, an alternative qPCR assay was developed to measure directly the DNA of G. graminis var. tritici. The assay was shown to detect DNA of G. graminis var. tritici in both symptomatic and non-symptomatic wheat roots, with an increase in the amount of DNA detected having a strong relationship with an increase in take-all symptoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Take-all of wheat and other cereals is caused by Gaeumannomyces graminis var. tritici, and to a lesser extent by the related fungus G. graminis var. avenae (e.g., Bithell et al. 2012b; Cook 2003). Gaeumannomyces graminis var. tritici infects roots of susceptible plants, resulting in black necrotic lesions. In severe cases, lesions spread to the stem bases leading to premature ripening of the affected plants (white heads) and a reduction in grain yield and quality. Losses of up to 60 % have been associated with G. graminis var. tritici infection of wheat in the United Kingdom (McMillan et al. 2011).
Take-all is commonly assessed in the field by visual inspection of plants. Symptoms similar to take-all on wheat roots can be caused by micro-organisms other than G. graminis var. tritici or by environmental conditions, however, leading to inconclusive identifications. Gaeumannomyces graminis var. tritici also lacks an active spore state on cereal hosts, which complicates microscopy-based diagnoses of this pathogen (Okubara et al. 2005). Consequently, time-consuming isolation of the pathogen and characterisation of fungal cultures have been required to confirm the presence of G. graminis var. tritici (Herdina et al. 1996).
While isolation and characterisation of fungal cultures is valuable for determining severity of infections and likely effects on yield, the measurement of pathogen DNA on plants or plant residues in the soil has proven less time-consuming and more quantitative. It can also provide greater understanding of the dynamics of inoculum multiplication in hosts or host tissues where the relationships between disease symptoms and inoculum are likely to be poor. For instance, take-all symptoms are seldom seen on rye (Secale cereale), but post-harvest concentrations of G. graminis var. tritici in soil can be considerably greater after rye than after wheat (Bithell et al. 2011). Indeed, quantitative PCR (qPCR) has been used to study pathogen incidence, dynamics and distribution in plant tissues during the course of infection of wheat by Fusarium spp., Rhizoctonia cerealis and Tapesia spp. (Okubara et al. 2005).
DNA-based assays exist to detect and quantify G. graminis var. tritici. For example, Herdina et al. (1996) developed a slot-blot hybridization technique for the semi-quantitative detection of Gaeumannomyces graminis var. tritici in infected roots and soil. Herdina and Roget (2000) also developed a quantitative DNA soil assay for detection of G. graminis var. tritici, but the assay had a low level of background signal particularly when DNA had been extracted from soils with high levels of organic matter. This was predicted to be due to non-specific DNA hybridization with the probe.
A DNA-based assay has been subsequently employed to estimate inoculum of G. graminis var. tritici and G. graminis var. avenae in soil and plant tissue (Ophel-Keller et al. 2008). This assay measures both the combined amount of inoculum DNA for these Gaeumannomyces species and also G. graminis var. avenae DNA alone. The amount of G. graminis var. tritici DNA is then extrapolated by subtracting the quantity measured for G. graminis var. avenae from the quantity calculated for both species. Use of the assay has demonstrated that G. graminis var. avenae is uncommon in the soils of New Zealand wheat crops (Bithell et al. 2012b). Furthermore, the assay has provided insights into the relationship between G. graminis var. tritici soil inoculum and disease severity, leading to its commercial use in New Zealand to predict the risk of severe take-all before sowing wheat crops (Bithell et al. 2012a). Nevertheless, the DNA-based diagnostic assay for detection of Gaeumannomyces spp. requires the use of multiple PCRs to extrapolate estimated amounts of G. graminis var. tritici rather than specifically measuring G. graminis var. tritici DNA. Furthermore, the commercialisation of the qPCR as a component of the PreDicta B test used to predict the risks of take-all in wheat crops prior to planting, can be prohibitive to its use due to the high associated costs.
This study describes the development of a qPCR-based assay to detect G. graminis var. tritici DNA in plant tissues. The qPCR can be used to quantify G. graminis var. tritici in the roots of plants with varying degrees of take-all to identify relationships between inoculum build-up and the expression of disease symptoms.
Materials and methods
Fungal isolates and culturing
Isolates of G. graminis var. tritici, G. graminis var. avenae and other fungi were obtained from collections or researchers in New Zealand and overseas, as described in Table 1. These fungi were selected to represent species of Gaeumannomyces closely related to G. graminis var. tritici on wheat and where possible fungi associated with cereal cropping systems or with grasses. All isolates were routinely grown on potato dextrose agar (PDA) medium at 25 °C. For long-term storage, isolates were kept on PDA slopes at 4 °C.
DNA extraction from fungi
To extract genomic DNA from fungi, each isolate was grown in a Petri plate on PDA overlaid with cellophane (Julzar Pty Ltd), until the plate was covered by freshly growing mycelia. The mycelia were then scraped off the cellophane and genomic DNA extracted using the PureGene™ Plant Tissue DNA Purification Kit (Nalgene) following the manufacturer’s instructions. DNA from each isolate was re-suspended in 200 μL of elution buffer (EB) (Qiagen). PCR was performed using the universal eukaryote primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (White et al. 1990) to confirm that the DNA could be used for PCR amplification. Reactions were performed in a total volume of 25 μL. The final concentrations of components were: 300 nM of each primer (Sigma-Aldrich), 20 mM Tris–HCl (pH 8.4), 1.5 mM MgCl2, 0.2 mM each dNTP and 1U Taq DNA polymerase (Invitrogen). Thermal cycling was performed in a GeneAmp PCR system 9700 as follows: initial denaturation at 94 °C for 2 min, followed by 35 cycles at 94 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min, with a final extension step at 72 °C for 7 min.
Design of G. graminis var. tritici-specific PCR primers
The translation elongation factor 1-alpha (EF1-α) gene was targeted for the design of primers to detect G. graminis var. tritici specifically. A 368-bp partial DNA sequence of EF1-α was amplified from each of eight isolates of G. graminis var. tritici (described in Table 1) using primers EF1-728 F (5′-CATCGAGAAGTTCGAGAAGG-3′) and EF1-986R (5′-TACTTGAAGGAACCCTTACC-3′) (Carbone et al. 1999). PCR reactions were conducted as described above for amplification of the ITS region. Thermal cycling was also performed as above, with the exception that the annealing temperature was 50 °C. PCR products were purified using a QIAquick PCR purification kit (Qiagen), and then sequenced in both directions with the appropriate primers (described above) (Macrogen Inc.). The EF1-α sequence for each strain was submitted to GenBank (accession numbers: JQ713812 to JQ713819).
The EF1-α DNA sequences for the eight selected G. graminis var. tritici isolates were compared with the EF1-α sequences from other Gaeumannomyces spp. as well as those of taxonomically related fungi and common wheat pathogens using ClustalW v1.83 (Thompson et al. 1997). From the resulting alignments of the EF1-α sequences, primers GgtEFF1 (5′-CCCTGCAAGCTCTTCCTCTTAG-3′) and GgtEFR1 (5′-GCATGCGAGGTCCCAAAA-3′) and a Taqman probe (GgtEFPR1, 5′-ACTGCACAGACCATC-3′) were designed using Primer Express (Applied Biosystems) to target G. graminis var. tritici specifically. The fluorogenic MGB probe was labelled at the 3′-end with a non-fluorescent quencher and at the 5′-end with the fluorescent reporter dye 6-FAM (6-carboxy-fluorescein) (Applied Biosystems).
Quantitative PCR
Amplification of the G. graminis var. tritici-specific 106-bp EF1-α fragment was performed using qPCR in a total reaction volume of 20 μL. The reaction mix included 10 μL of 2× iTaq Supermix with Rox (BioRad Laboratories). The MGB probe GgtEFPR1 was added to a final concentration of 0.15 μM and the primers GgtEFF1 and GgtEFR1 to a final concentration of 0.3 μM. One hundred ng of template DNA was then added to each reaction. PCR amplifications were carried out in a StepOnePlus (Applied Biosciences) with the following steps: (i) initial denaturation at 95 °C for 3 min, (ii) 40 amplification cycles of 94 °C for 15 s, 52 °C for 20 s, and 72 °C for 20 s. Quantities of target DNA were estimated using a standard curve constructed by regressing Ct values onto the log10 of the concentration of G. graminis var. tritici DNA standards. The standards were produced by 10-fold serial dilutions of genomic DNA from an initial concentration of 100 ng/μl. The regression equation was used to calculate estimated amounts of G. graminis var. tritici target DNA in each sample. Each DNA sample and the DNA standards were amplified in duplicate, and negative and positive controls were included with each PCR assay.
Collection of plant samples and take-all assessment
Wheat plant samples at various growth stages (GS) (Zadoks et al. 1974) were collected from Chertsey in New Zealand. For each growth stage, approximately five plants were randomly selected from each of 20 locations in the crop, with locations evenly spaced on a W transect that covered the width of the paddock. The roots of each plant were washed and assessed for take-all severity on a scale of 0 to 4 (Category 0 = no take-all; Category 1 = 1 to 10 % of the root system with black necrotic lesions; Category 2 = 11 to 30 %; Category 3 = 31 to 60 %; Category 4 = 61 to 100 %). For post-harvest samples (GS 87), scoring was difficult because of degradation of senesced roots, so take-all symptoms were scored as ‘none’ (Category 0), ‘some’ (Category 1–3) or ‘severe’ (Category 4). The amounts of G. graminis var. tritici DNA in the roots of 2–4 plants in each take-all category at each time point were subsequently quantified by qPCR.
Extraction of total DNA from wheat roots
For extraction of total DNA from wheat roots, the entire root system of a plant was crushed in a mortar with liquid nitrogen. Approximately 100 mg of the powdered root tissue was added to a screw cap vial containing 800 μL AP1 lysis buffer (Qiagen) and 1 g of 2.3-mm steel beads. Bead-beating was performed for 1 to 3 min to macerate the root tissue and DNA was extracted using the DNeasy Plant Minikit (Qiagen) following the manufacturer’s instructions. Total DNA was re-suspended in 200 μL of EB (Qiagen) and stored at −80 °C before use. A sample of total DNA was extracted from each crushed root system in duplicate.
Each sample was diluted 10-fold prior to qPCR. To identify PCR inhibitors, a Sybr green-based qPCR was performed using the primers Pot25SF (5′-TCGGAATTCGAAGCTAGAGG-3′) and Pot25SR (5′-AACGTCGCTATGAACGCTTG-3′) and the diluted DNA as a template. Pot25SF and Pot25SR were designed as part of this study, to target the plant 25S ribosomal RNA gene sequence. Reactions were performed in a total volume of 20 μL including 10 μL of 2× iTaq Sybr Green with Rox (BioRad Laboratories) and a final concentration of 0.3 μM of each primer. The same PCR protocol described above for the G. graminis var. tritici-specific qPCR was used for amplification of the 25S ribosomal DNA sequence, with the exception that an annealing temperature of 60 °C was used.
Results
The primer pair GgtEFF1 and GgtEFR1 and the Taqman probe GgtEFPR1 were designed to amplify a G. graminis var. tritici-specific EF1-α fragment of 105 bp. The primer and probe sequences were 100 % identical to the complementary DNA sequences for all G. graminis var. tritici isolates. The GgtEFF1 primer had three nucleotide mismatches to the EF1-α sequence for G. graminis var. avenae, whereas the primers and probe had nine nucleotide mismatches to the EF1-α sequence for G. cylindrosporus (Fig. 1).

Nucleotide sequence alignment of the partial EF1-α sequences from Gaeumannomyces graminis var. tritici (Ggt), G. graminis var. avenae (Gga) and G. cylindrosporus. The alignment shows the location of the primers GgtEFF1 (dark green arrow) and GgtEFR1 (light green arrow) and the Taqman probe GgtEFPR1 (red arrow). This primer and probe combination was designed for the specific detection of G. graminis var. tritici. Coloured squares show polymorphic nucleotides on the divergent sequences. A dash represents a missing nucleotide in the associated EF1-α sequence. The primers were designed to amplify a 105-bp fragment
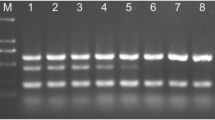
The specificity of the GgtEF primer and probe set was initially verified by performing qPCR on DNA from a collection of G. graminis var. tritici isolates. Other closely related species (e.g., G. graminis var. avenae and G. cylindrosporus) were also tested as well as fungi typically found growing in the same environments (e.g., Fusarium graminearum) (see Table 1). In these assays, only G. graminis var. tritici isolates generated an amplicon (Table 1). An ITS amplicon was produced by all DNA samples using the universal primers ITS1 and ITS4 demonstrating that PCR products could be amplified from all the DNA templates (data not shown).
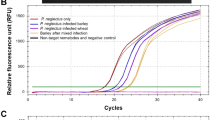
The efficiency of the qPCR and the theoretical limits of detection for the assay were established by performing qPCRs on a 10-fold serial dilution of genomic DNA extracted from G. graminis var. tritici. The amplification curves (e.g., Fig. 2) showed that the G. graminis var. tritici-specific qPCR had an R2 = 0.96 to 0.99 and a Y-intercept of approximately 35.57. Gaeumannomyces graminis var. tritici DNA was quantifiable to a minimum of 1 pg. Similar results were obtained for the calibration samples when they were used in later qPCR runs, which were carried out to quantify G. graminis var. tritici DNA in field-collected plant material. Although quantities of DNA below 1 pg could be detected, these lay outside the linear range for detection, so that accurate quantification of G. graminis var. tritici in this range was not possible. Instead, positive qPCR results for these smaller quantities of G. graminis var. tritici DNA were used only to indicate the possible presence of the pathogen.

Standard curve for the quantitative detection of Gaemannomyces graminis var. tritici DNA using primers GgtEFF1 and GgtEFR1 in conjunction with the Taqman probe GgtEFPR1. Black dots represent data used to establish the linear range of the qPCR. Results of qPCR are given as Cycle threshold (Ct). Those samples that failed to produce a G. graminis var. tritici amplicon by a Ct of 40 were recorded as ‘undetermined’. Gaeumannomyces graminis var. tritici was considered not detected in samples recorded as undetermined
To validate the G. graminis var. tritici qPCR assay as a tool to measure the relationships between the quantity of G. graminis var. tritici DNA detected in wheat roots and the symptoms of take-all observed in the field, plants with a range of disease categories between 0 (no take-all) and 4 (severe take-all) were tested for G. graminis var. tritici DNA. Roots with increasing severity of take-all generally showed increasing quantities of G. graminis var. tritici DNA regardless of the growth stage of the assayed plants (Spearman’s rank correlation r = 0.56; p < 0.001; Fig. 3). For example, wheat roots with a disease category of 4 showed the greatest quantities of G. graminis var. tritici DNA (up to 9.4 × 104 ng/g root tissue), on average five-fold greater than roots with a take-all severity score of 3. The quantities of G. graminis var. tritici DNA were, however, on average similar in samples with a disease category of 1 or 2. Gaeumannomyces graminis var. tritici DNA was detected in a number of root samples collected at different times with a disease severity category of 0, although quantities were less than in plants with visible take-all symptoms.

Estimated quantities of Gaeumannomyces graminis var. tritici DNA (ng/g root tissue) in roots of wheat plants with increasing take-all (categories 0 to 4), which were collected at different growth stages (GS) from Chertsey, New Zealand. Large symbols: means for each category. Small symbols: individual data values
Discussion
Quantitative diagnostic assays are important tools in epidemiological studies of plant diseases as they enable the temporal and spatial monitoring of fungal populations. In the present study, a protocol was developed for the quantitative detection of G. graminis var. tritici DNA from wheat roots. The specificity of the assay was demonstrated by successful differentiation of G. graminis var. tritici from related Gaeumannomyces such as G. graminis var. avenae as well as other taxonomically related fungi. It was also sensitive, detecting pathogen DNA to below 1 pg in each PCR reaction. A DNA-based assay already exists to quantify G. graminis var. tritici DNA in environmental samples (Ophel-Keller et al. 2008). The assay has been used as a predictive tool for take-all in wheat crops (Bithell et al. 2012a), but its commercialisation and the associated costs limit its use for many researchers and it doesn’t detect the pathogen DNA directly. Thus, our qPCR assay provides an alternative method for the detection of G. graminis var. tritici.
A strong relationship was observed between increasing quantities of G. graminis var. tritici DNA in roots and take-all disease progression. The quantities of G. graminis var. tritici DNA in roots of low severity Category 1 and Category 2 plants were similar, however. This may be due to the ectotrophic growth habit of this fungus in the early stages of root colonisation. Ectotrophic expansion of G. graminis var. tritici along the root segments would be unlikely to result in disease symptoms, yet qPCR would still detect the presence (and an increase) of pathogen DNA. Alternatively, this may result from an inability to discriminate visual symptoms for these less severe disease categories. The relative variation in the quantities of G. graminis var. tritici DNA from plants in Categories 1 and 2 was greater than for more severely affected plants.
Assigning plants within the take-all category range 0–2 was difficult late in the growth season due to their roots having already senesced. Nevertheless, there were large differences in the quantities of G. graminis var. tritici DNA detected in plants belonging to these categories. Perhaps senescing and senesced roots do not react to invasion by G. graminis var. tritici and, similarly, already blackened roots do not visibly change as inoculum further increases or degrades. The differentiation of inoculum in senescent tissue by qPCR will support future studies of G. graminis var. tritici inoculum dynamics in dead or dying tissue where take-all symptoms are hard to define. Traditionally such studies have relied on laborious isolation and culturing techniques.
Conclusion
In summary, a qPCR assay was developed that related increasing disease symptoms with increasing quantities of G. graminis var. tritici DNA in wheat roots during take-all epidemic development. Disease assessment, however, only reflects inoculum concentrations in roots of growing plants. Thus, use of qPCR also provided a novel tool for evaluating inoculum of G. graminis var. tritici in roots of non-symptomatic plants as well as in senescent plant material. Further work is planned to examine the build-up in inoculum on roots during host plant senescence and beyond.
References
Bithell SL, Butler RC, Harrow S, McKay A, Cromey MG (2011) Susceptibility to take-all of cereal and grass species, and their effects on pathogen inoculum. Ann Appl Biol 159:252–266
Bithell SL, McKay A, Butler RC, Herdina, Ophel-Keller K, Hartley D, Cromey MG (2012a) Predicting take-all severity in second-year wheat using soil DNA concentrations of Gaeumannomyces graminis var. tritici determined with qPCR. Plant Dis 96:443–451
Bithell SL, McKay A, Cromey MG (2012b) Low frequency of Gaeumannomyces graminis var. avenae in New Zealand; implications for take-all management in wheat. Australas Plant Pathol 41:173–178
Carbone I, Anderson JB, Kohn LM (1999) Patterns of descent in clonal lineages and their multilocus fingerprints are resolved with combined gene genealogies. Evolution 53:11–21
Cook RJ (2003) Take-all of wheat. Physiol Mol Plant Pathol 62:73–86
Herdina, Roget DK (2000) Prediction of take-all disease risk in field soils using a rapid and quantitative DNA soil assay. Plant Soil 227:87–98
Herdina, Harvey HP, Ophel-Keller K (1996) Quantification of Gaeumannomyces graminis var. tritici in infected roots and soil using slot-blot hybridization. Mycol Res 100:962–970
McMillan VE, Hammond-Kosack KE, Gutteridge RJ (2011) Evidence that wheat cultivars differ in their ability to build up inoculum of the take-all fungus, Gaeumannomyces graminis var. tritici, under a first wheat crop. Plant Pathol 60:200–206
Okubara PA, Schroeder KL, Paulitz TC (2005) Real-time polymerase chain reaction: applications to studies on soilborne pathogens. Can J Plant Pathol 27:300–313
Ophel-Keller K, McKay A, Hartley D, Herdina, Curran J (2008) Development of a routine DNA-based testing service for soilborne diseases in Australia. Australas Plant Pathol 37:243–253
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTALX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
White T, Bruns T, Lee S, Taylor J, Innis M, Gelfand D, Sninsky J (1990) PCR protocols: a guide to methods and applications. Academic, New York
Zadoks JC, Chang TT, Konzak CF (1974) A decimal code for the growth stages of cereals. Weed Res 14:415–421
Acknowledgments
The authors thank Richard Falloon and Simon Bulman for their reviews of the manuscript. This work was funded by New Zealand’s Ministry for Business, Innovation and Employment through programme LINX0804.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Keenan, S., Cromey, M.G., Harrow, S.A. et al. Quantitative PCR to detect Gaeumannomyces graminis var. tritici in symptomatic and non-symptomatic wheat roots. Australasian Plant Pathol. 44, 591–597 (2015). https://doi.org/10.1007/s13313-015-0379-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13313-015-0379-y