
Abstract
Popular is not only an important tree species economically, but is also a model system for plant biology. Marssonina leaf spot is a serious leaf disease caused by Marssonina brunnea, which can result in significant economic losses. M. brunnea is considered to be a hemibiotrophic pathogen but its pathogenesis remains unclear. Full length cDNA of 24 LysM effector genes were obtained based on the sequencing of the M. brunnea genome. All of these genes belonged to a single gene family for which the characteristics of the amino acid sequences and protein domains were analyzed. Further detailed analysis was carried out on two representative gene family members. These two genes were up-regulated during the infection process and, when transformed into Arabidopsis thaliana, reduced the plant’s sensitivity to chitin thus might compromised its immune system. LysM effectors appear to show a specific affinity for chitin since the expressed proteins co-precipitated with insoluble chitin. We performed a series of studies into the molecular mechanism of LysM effectors from M. brunnea and carried out a preliminary analysis into the interaction between the pathogen and its host. This research lays a foundation for further investigation into forest tree resistance breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Black spot disease, caused mainly by Marssonina brunnea, is a destructive fungal disease of poplar (Spiers 1983). When poplars are infected, punctiform black spots appear on the leaves and these then become enlarged resulting in premature defoliation. Consequently, the life cycle of the infected poplar is greatly shortened, together with a rapid reduction in the production of biomass (Wu et al. 1992). Since the Populus genus is the most important tree genus economically for this country, black spot disease has caused enormous losses over the last few decades.
Currently, few effective methods can prevent or control black spot disease. Chemical methods are restricted by the lack of suitable antifungal agents, whilst conventional disease-resistance breeding has limitations due to the low efficiency of selection and long breeding period. Hence, there is a need for molecular biology-based applications to be developed. In order to do this, a better understanding of the molecular mechanisms is required.
M. brunnea is a pathogenic hemibiotrophic fungus and as such is biotrophic, growing first on living tissue, and then later, necrotrophic, getting its nutrients from dead tissues by killing the host plant. M. brunnea displays a relatively long infection period before it finally kills the tissue. During this process, pathogen-associated molecular patterns (PAMPs) from the pathogenic fungus are recognized by Populus, eliciting the pathogen-triggered immunity (PTI) of host (Jones and Dangl 2006). Hence, M. brunnea also needs to take measures to suppress the host’s defense reactions to enable further invasions. An effective strategy for doing so is for the pathogenic fungi to secrete small effector proteins that may aid the pathogen in evading recognition by the plant. Effectors play a crucial role in blocking signal transmission or can even directly suppress the expression of defense-related genes. Much of the research into plant immunity has focused on effectors, however, it still remains in the early stages and only a few effectors have been identified so far (Giraldo and Valent 2013).
To discover more potential effectors, we previously isolated several secretory proteins from M. brunnea and performed full length cDNA cloning. The results showed that, of all the secretory proteins obtained, nine contained the same identical domain, the so-called LysM domain, and so were considered to belong to the same protein family (Cheng et al. 2010). Further investigation of the whole genome sequence of M. brunnea revealed the existence of several more family members and moreover, that this gene family is the largest one in M. Brunnea (Zhu et al. 2012).
Transcriptome analysis showed that the expression of these LysM genes tended to increase during the infection stage, indicating that they may be related to the pathogenicity of M. Brunnea (Zhu et al. 2012). Currently, LysM domains are known to exist in bacteria, fungi, animals and plants. These domains (Pfam PF 01476) range in length from 44 to 65 amino acids residues and recognize peptidoglycan and chitin. (Buist et al. 2008). Research on Cladosporium fulvum has shown that LysM domains may weaken host cell defense responses (de Jonge et al. 2010).
In order to further investigate the roles of the LysM domain in pathogen-host interaction, we identified 24 members of this gene family and then selected two representative genes for functional verification.
Materials and methods
Materials
M. brunnea f. sp. multigermtubi was isolated from Populus canadensis and streaked onto both potato dextrose agar (Sigma, USA) and simple liquid media (2 % w/v sucrose, 0.5 % w/v yeast extract) for subculturing (Cheng et al. 2010). The infection assay was performed as described (Cheng et al. 2010). the mycelia were collected with filter paper, frozen rapidly using liquid nitrogen and stored at −80 °C for later extraction of DNA and RNA.
Branches of P. Canadensis were taken from biennial cuttage seedlings and cultured in the greenhouse at 25 °C and 70 % relative humidity under a 16 h/8 h photoperiod.
Wild-type A. thaliana Col-0 were cultured in a constant temperature incubator at 23 °C under a 16 h/8 h light/dark cycle at 60 % relative humidity with supplemental lighting (5000 lux).
Full-length cDNA cloning
RNA was extracted from the mycelia of M. brunnea using a modified hexadecyltrimethylammonium bromide (CTAB) method (Jiang et al. 2011). First-strand cDNA was synthesized using 3’-Full RACE Core Set Ver. 2.0 (Takara, Japan) and 5’-Full RACE Kit according to the manufacturer’s instructions (Takara, Japan), and then used as template for 3’RACE and 5’RACE. The PCR fragments were ligated into the pMD19-T (Takara, Japan) vector for sequencing. The full-length cDNA sequence was acquired by splicing the obtained fragments with the aid of Bioedit and these cDNA fragments were inserted into the pMD19 simple Vector for sequencing (Takara, Japan). Multiple sequence alignment was constructed using ClustalX2. Analysis of the molecular weight and isoelectric point (pI) of the proteins and their amino acid composition was performed using ProtParam (http://web.expasy.org/protparam/) The Pfam database was used to predict protein domains (http://pfam.sanger.ac.uk/). Predictions of signal peptides were made using SignalP (http://www.cbs.dtu.dk/services/SignalP) .
qRT-PCR Analysis
A modified CTAB method was used for the extraction of total RNA from poplar and A. thaliana (Xu et al. 2009). Total RNA was first treated with DNase I (Takara, Japan) to remove any genomic DNA contamination and then reverse-transcribed into cDNA using PrimeScript™ RT-PCR kit (Takara, Japan). The cDNA was diluted 1:3 with nuclease-free water to create the qRT-PCR template.
Reactions were carried out in a 20 μl reaction volume consisting of the following components: 10 μl 2 × SYBR Premix Ex TaqTM (Takara, Japan); 0.4 μl 10 μM PcCHI-f primers; 0.4 μl 10 μM PcCHI-r, 0.4 μl ROX reference dyeII; 2 μl cDNA template and 6.8 μl Milli-Q water. qRT-PCR was performed on an Applied Biosystems 7500 Real-time PCR system with the following cycling conditions: 95 °C for 30 s, 95 °C for 5 min and 60 °C for 34 s for 40 cycles. A dissociation curve was analyzed after the reaction to exclude the possibility of nonspecific amplification. Three replicates were performed for each reaction.
Primers for semi quantitative analysis were listed in Table S1, and beta-actin was chosen as an internal control gene (Morris et al. 2010) . Several defense-related genes were analyzed (Hamamouch et al. 2011; Czechowski et al. 2004; Galletti et al. 2011; Bi et al. 2010).
Protein expression
BL21 strain was used to perform the prokaryotic expression. Positive microbial suspensions were obtained after shaking at 23 °C for 24–30 h, then the suspensions were diluted in a ratio of 1:40 into 2 ml LB media (LB media: Tryptone 10 g L−1, Yeast extract 5 g L−1, NaCl 5 g L−1; Ampicillin 100 μg mL−1, Chloromycetin 35 μg mL−1). BL21 (DE3) plysS was used as a negative control. The microbial suspension was then diluted to 20 ml and incubated at 23 °C with shaking until the optical density at 600 nm (OD600) reached 1.5–1.6. IPTG was added (final concentration 0.4 mM) and the cells were incubated at 28 °C for 2 h. After this time, the microbial suspension was poured into a 50-ml centrifuge tube, centrifuged and the supernatant removed. Phosphate buffered saline (5 ml) (NaCl 8.0 g L−1, KCl 0.2 g L−1, Na2HPO4 1.44 g L−1, KH2PO4 0.24 g L−1, pH 7.4, 1%Triton X-100,1 mM PMSF) was added to resuspend the cells and this was then cleared by sonication. Total proteins were isolated by centrifuging the cell suspension at 8000 × g for 20 min at 4 °C and then filtering off the supernatant containing the protein extract. Loading buffer (2.5 % w/v SDS, 25 % glycerol, 125 mM Tris-Cl (pH 6.8). 0.01 % w/v bromophenol blue) was added to the samples, which were then heated to 95 °C for 5 min. The samples were stained with R-250 for 5–10 min and destained with 10 % acetic acid. The results were observed, scanned and saved as photo files. Proteins from the prokaryotic expression were gently vortexed and incubated with insoluble chitin (Sigma USA) at room temperature for 3 h and then centrifuged at 13,000 × g for 5 min. The supernatant was discarded and the sediment was rinsed three times with sterile water. Following this, 1 % sodium dodecyl sulfate (SDS) was added to the samples before being boiled for over 5 min. The supernatant was then used for Tricine SDS Polypropylene amide gel electrophoresis (PAGE). Separating gel with 8 % acylamide percentage was used. The total running time should be about 1 h for a 120 V voltage. Western blotting was performed as described using GST antibody (Burnette 1981).
Vector construction and plant transformation
A signal peptide of PR1a was added before LysM genes using overlapping PCR and constructed into binary vector PH35GS to express a fusion peptide which can secreted to intracellular space (Mitsuhara et al. 2000). The resulting vector was transformed into Agrobacterium tumefaciens strain EHA105 and then used to transform A. thaliana using a floral dip as previously described (Clough and Bent. 1998). After transformation, the wide-type plants and transgenic plants were both grown in the same environment.
Chitin treatment of A. thaliana
Chitooligosaccharides (tetrachitooligosaccharides, pentachitooligosaccharides and hexachitooligosaccharides) were purchased from the Megazyme Company (Germany). Solutions at a final concentration of 0.1 μM, 1 μM, 5 μM and 10 μM were prepared using sterile water. These were sprayed on comparable mature, intact A. thaliana leaves. Samples were taken at different timepoints (0 h, 0.5 h, 1 h, 1.5 h, 2 h) post application, rapidly frozen and stored in liquid nitrogen.
Results
Cloning of LysM gene family members from M. brunnea
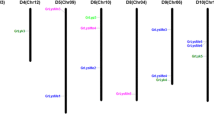
The full length cDNA of the LysM gene family ranged from 690 bp to 1179 bp, with most of them being around 800 bp. The coding amino acid sequence was around 150 aa and the molecular weight ranged from 15.52 kDa to 16.67 kDa. All of the coding proteins contained an 18 bp or 19 bp signal peptide for protein secretion, and also known as a common feature of fungal effector proteins (Table 1). Eighty-nine scaffolds were acquired through the whole genome sequencing of M. brunnea, while LysM genes were distributed on 18 of them. We renamed all LysM genes according to their distribution.
The pI of the LysM effectors from M. brunnea vary greatly from those in prokaryotes. The theoretical pI of the M. brunnea effector proteins ranged from 4.34 to 8.54 with most of them being less than 5.5 (Table 1). Only MbLysM03, MbLysM04, MbLysM10, MbLysM20 and MbLysM24 had a pI over 7 and for MbLysM23 the pI was between 6 and 7. In contrast, in prokaryotes the pI of the LysM domain proteins ranged from 4 to 12, with most being between 5 and 10, notably within the interval from 8 to 10 (Buist et al. 2008). These results may reflect the innate differences between eukaryotes and prokaryotes.
Comparison of M. brunnea LysM effector family members
Multiple alignments showed that different members of this gene family had relative high homology (Fig. S1A). Two LysM domains were found in all family members based on the domain analysis of amino acid sequences. The first, LysM I, was located towards the middle of the sequence and its length was generally 41 amino acids. The second domain, LysM II, was found at the C-terminal of the proteins and was longer than LysM I with more variation in its length, ranging from 44 to 47 amino acids. The conserved region of the two LysM domains appeared within the first half of the domain sequence. Further analysis of the domain sequences revealed the presence of some highly conserved sites including the first amino acid, Tyr (Y); the 15th, Asp (D); the 17th, Cys (C) and the 28th Asn (N). Besides these, the second, seventh, 16th, 47th and 48th were also reasonably well conserved (Fig. S1B).
Domain analysis and expression of two LysM genes from M. brunnea
Two LysM genes from M. brunnea were selected for further detailed analysis. They were named MbLysM5 and MbLysM19 (Fig. 1A). These two genes encoded secretory proteins which had been detected by us before (Cheng et al. 2010). The full length cDNA of these two genes were 1179 bp and 962 bp, respectively, which encoded proteins of 143 and 145 amino acids, respectively. Further sequence analysis showed that the pI (5 and 4.84) of the two proteins and their amino acid compositions were similar, with Ala, Asp, Leu and Thr comprising a large proportion of all the amino acids present. The LysM II domains at the C-terminal of both proteins were conserved. However, the LysM I domains which located in the middle of these two proteins are different. The LysM I domain of MbLysM5 is a integrate LysM domain, whereas the LysM I domain in MbLysM19 only aligned with the first 28 amino acids of a typical LysM domain (Fig. 1B).

Domain analysis, sequence alignment and expression patterns of two LysM genes in Marssonina.brunnea. a. Signal peptide and LysM domains in LysM genes; b. Sequence alignment of LsyM domains bwtween two LysM genes; c. Expression analysis of Marssonina.brunnea LysM genes during infection. Four time points (12hpi, 24hpi, 48hpi, 96hpi) were selected
We inoculated poplars with M. brunnea to investigate the expression patterns of the M. brunnea LysM genes in the infection process. Four time point, 12 h, 24 h, 48 h and 96 h after inoculation were selected for qRT-PCR analysis (Fig. 1C). As the results shown, the expression of both genes was relatively stable without any remarkably change in the early stages, but had a significant up-regulated at 96 h after inoculation. It indicated that these two genes might come into play a role during this stage. Furthermore, the expression patterns of MbLysM5 and MbLysM19 were similar to each other, which indicate the same function they might have..
LysM effectors can bind with chitin in vitro
To find the possible relationship between chitin and LysM effectors. We constructed recombinant vectors to express MbLysM19-GST and MbLysM5-GST fusion proteins using prokaryotic expression system. Total proteins were extracted and then detected by SDS PAGE. With expectation, a band between 33 kDa and 45 kDa was detected. BL21 (DE3) plysS was used as a negative control and no bands were detected (Fig. 2A). We then mixed the fusion proteins with insoluble chitin. After 3 hours incubation, the supernatant was discarded, and the sediment was washed for three times. Then the sediment was boiled and analyzed by SDS PAGE. As the results shown, the band with correct size was still visible indicating that the fusion proteins were capable of binding to chitin (Fig. 2B). Western blotting assays was performed to confirm that the proteins binding to chitin were the LysM-GST fusion proteins (Fig. 2C).

Prokaryotic expression of LysM-GST fusion protein and their binding with insoluble chitin. a. Prokaryotic expression of MbLysM19-GST and MbLysM5-GST fusion proteins; b. The fusion protein co-precipitate with insoluble chitin; c. Western blotting assays confirmed the protein co-precipitate with insoluble chitin were MbLysM19-GST and MbLysM5-GST fusion proteins
Resistance-related genes in A. thaliana are induced under chitin treatment
Using the housekeeping gene beta-actin as a reference genes, we performed semi-quantitative analysis on known A. thaliana early defense genes (PR1, PR2, PDF1.2, WRKY22, WRKY29 and FRK1). All of these defense genes, except PR2, showed increased expression levels. Among them, PDF1.2 showing the most significant change (Fig. 3A).

The defense-related genes of A.thaliana were induced by chitooligosaccharides.a. Expression of seveal defense-related genes (PR1, PR2, PDF1.2, WRKY22, WRKY29, FRK1) induced by chitooligosaccharides in A.thaliana. b. Expression of PDF1.2 gene induced by chitooligosaccharides with different concentration (0 μM, 0.1 μM, 1 μM, 5 μM and 10 μM); c. Expression of PDF1.2 gene induced by chitooligosaccharides with different polymerization degree. CHI4 (tetra chitooligosaccharides), CHI5 (pentachitooligosaccharides) and CHI6 (hexachitooligosaccharides); d. Expression of PDF1.2 gene treated with chitooligosaccharides for different time (0 h, 0.5 h, 1 h, 1.5 h and 2 h)
The expression level of PDF1.2 increased concurrently with in the chitooligosaccharides concentration until it arrived the peak when 5 μM chitooligosaccharides was used. No further changes in expression were observed even using 10 μM chitooligosaccharides for inducement (Fig. 3B). The degree of chitooligosaccharides polymerization also affected the induction. We treated A. thaliana with chitooligosaccharides with three types of polymerization degree. Although all resulted in the increased expression of PDF1.2, the upregulation was much more significant when treated with hexachitoologosaccharides (Fig. 3C). In A. thaliana, 0.5 h after treatment with chitoologosaccharides, increases in the expression levels of PDF1.2 could be observed. As the incubation time increased, the expression level of PDF1.2 also increased gradually, reaching its peak of expression at around 1.5 h, and decreasing slightly after this time point (Fig. 3D)..
LysM effectors weaken chitin-induced defense responses in A. thaliana
As described above, a possible mechanism for LysM effectors may come from their binding to chitin to avoid recognition by the host. Hence, they would be expected to be localized in the extracellular matrix. To investigate this, we added a tobacco PR1a signal peptide at the N-terminal of the LysM genes to aid the secretion of the LysM genes from the plant cells (Mitsuhara et al. 2000). We then re-construct vectors based on PH35GS, the recombinant plasmid PH35GS-MbLysM19 and PH35GS-MbLysM5 were introduced into A. thaliana Col-0 using the floral dip technique (Clough 2005). The T1 generation seeds were screened with hygromycin in MS media and resistant seedlings were obtained. All these transformants was verified by PCR. These seedlings were transferred onto soil once four leaves had appeared. The seedlings were kept in normal growth conditions until they started flowering and were then treated with 5 μM hexa-chitooligosaccharides for 1.5 h.
Semi-quantitative PCR analysis showed that the expression levels of PDF1.2 only significantly increased after treatment with chitooligosaccharides in A. thaliana transformed with PH35GS. In comparison, A. thaliana transformed with MbLysM19-PH35GS and MbLysM5-PH35GS were insensitive to PDF1.2 induction by chitooligosaccharides, especially LysM5, which showed almost no change in PDF1.2 expression (Fig. 4). This indicates that the LysM genes suppressed the defense response of plants normally induced by chitoologosaccharides.

The expression of PDF1.2 gene with and without chitin treatment A.thaliana col0 was used as control. The PDF1.2 genes was obviously up-regulated when treated with chitooligosaccharides. But in the A.thaliana transformed with MbLysM19-PH35GS, the expression of PDF1.2 was only slightly increased and in A.thaliana transformed with MbLysM5-PH35GS, the expression of PDF1.2 was even not changed
Discussion
Pathogenic fungi can be divided into three categories: biotrophic, necrotrophic and hemibiotrophic according to their nutrition acquisition methods. Biotrophs live in a parasitic style, acquiring their nutrition from the host’s living tissues. Necrotrophs kill the host first, and then decompose their tissues for survival. Hemibiotrophic fungi could also be regarded as necrotrophs but with weaker virulence, although they do also kill the hosts (Lee and Rose 2010). During a long-term infection process, they may additionally acquire nutrition from the host’s living tissues. M. brunnea belongs to this latter group. There is often a long period of time from the initial infection to the eventual death of the host, and during this period, pathogenic fungi face recognition by the host and the resulting immune response. As a result, pathogenic fungi have evolved effector proteins, which are released into the host and suppress the host’s defense reactions (Chisholm et al. 2006). Fungi have more complex genomes and a higher degree of evolution and thus possess a greater variety of means for infection. Hence, it is more difficult to perform functional analysis on fungal effectors than on those from bacteria. Until now, only a few fungal effectors have been identified, including the LysM effectors, which have a close relationship with the virulence of the pathogenic fungi. They are so-named because they only possess LysM domains. The LysM effectors exist widely in all types of fungi, although their function during infection is unclear. De Jonge and co-workers discovered that the LysM effector, ECP6, could bind to chitin (De Jonge et al. 2010). Since then, research on LysM effectors and chitin has been performed in other pathogenic fungi. However, previous studies have only managed to identify one or two genes from any one species, and there have been no reports on the identification and characterization of a whole LysM gene family.
We cloned a LysM effector gene family containing 24 genes, based on the analysis of the whole genome sequence of M. brunnea. The conserved regions of the M. brunnea LysM genes were mainly in the LysM domains. Some highly conserved sites were found within the LysM domains, which are likely to be essential for the stability of the domain structure and may contribute to chitin binding. Analysis on the expression patterns of the LysM genes showed that the highest expression was during the later stages of infection. Since it is known that chitinases decompose the cell walls of fungi generating a large amount of chitin fragments, we suggest that the chitinases of poplars are likely to perform their function during the late stage of infection. To prevent these chitin fragments from triggering the poplar immune system, M. brunnea releases LysM effectors that bind to the chitin fragments thus weakening the immune response of poplar.
Functional verification in A. thaliana proved that LysM effectors of M. brunnea could indeed suppress the expression of resistance-related genes which might be the downstream targets of chitin-induced signaling pathway. This confirmed that as fungal effectors, LysM proteins from M. brunnea can inhibit the PAMPs-triggered immunity. It also indicated that this suppression of immune responses might be universal for this group of effectors since it was not only restricted to poplars, but also occurred in A. thaliana.
The number of LysM domains within a protein may also affect its function. Among the LysM effectors reported previously, three LysM domains were found in ECP6 and Mg3LysM, two LysM domains in slp1, and only one in Mg1LysM (de Jonge et al. 2010; Marshall et al. 2011; Mentlak et al. 2012). These effectors can bind to chitin directly and the number of LysM domains has no effect on binding. However, Mg1LysM from Mycosphaerella graminicola, which possesses only one LysM domain (Marshall et al. 2011), could not suppress the chitin-induced defense responses and so it appeared that LysM genes containing two or more LysM domains were necessary for this function. Most of M. brunnea LysM genes have two LysM domains. However, of the two genes analyzed, MbLysM5 with two integrated LysM domains suppressed the host immunity more significantly than MbLysM19 with only one integrated LysM domain, indicating that the integration of the LysM domain is essential for LysM effectors to play a role.
LysM effectors in pathogenic fungi can weaken the host’s defense responses because they can compete with the chitin elicitor receptors in the host. CEBIP in rice is a typical LysM receptor (Shimizu et al. 2010; Hayafunea et al. 2014). Recent studies have revealed that the effector, slp1, from the rice blast pathogen Magnaporthe oryzae, which contains LysM domains, can compete with CEBIP to bind to chitin (Mentlak et al. 2012). The homolog of CEBIP exists in Populus and we believe that it may have a competitive relationship with one of the LysM effectors that we have identified. Furthermore, CERK and CEBIP of rice can form a dimer when elicited by chitin, however, it has not yet been shown whether dimerization also occurs in poplar, nor whether the LysM effectors of M. brunnea can form a heterodimer with CERK in poplars. The latter deserves further attention, since, if they can form a heterodimer, the CERK and CEBiP dimer will presumably be damaged, and thus the immunity response will be weakened.
The long-term interaction between M. brunnea and poplar means that they have co-evolved with new genes appearing to substitute for ones that had been recognized by each other. It should be a possible reason for the existence of so many LysM domain-containing genes with a similar sequence and structure in both species. Studies on such a gene family will lay a foundation for the understanding of co-evolution between pathogen and host.
References
Bi DL, Cheng YT, Li X, Zhang YL (2010) Activation of Plant Immune Responses by a Gain-of-Function Mutation in an Atypical Receptor-Like Kinase. Plant Physiol 153:1771–1779
Buist G, Steen A, Kok J, Kuipers OP (2008) LysM, a widely distributed protein motif for binding to (peptido) glycans. Mol Microbiol 68:838–847
Burnette WN (1981) “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112:195–203
Cheng Q, Cao Y, Jiang C, Xu L, Wang M, Zhang S, Huang M (2010) Identifying secreted proteins of Marssonina brunnea by degenerate PCR. Proteomics 10:2406–2417
Chisholm ST, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124:803–814
Clough SJ (2005) Floral dip: agrobacterium-mediated germ line transformation. Methods Mol Biol 286:91–102
Czechowski T, Bari RP, Stitt M et al (2004) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot- pecific genes. The Plant J 38:366–379
de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MH, Thomma BP (2010) Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329:953–955
Galletti R, Ferrari S, De Lorenzo G (2011) Arabidopsis MPK3 and MPK6 play different roles in basal and oligogalacturonide-or flagellin-induced resistance against Botrytis cinerea. Plant Physiol 157:804–814
Giraldo MC, Valent B (2013) Filamentous plant pathogen effectors in action. Nat Rev Microbiol 11:800–814
Hamamouch N, Li C, Seo PJ, Park CM, Davis EL (2011) Expression of Arabidopsis pathogenesis-related genes during nematode infection. Mol Plant Pathol 12:355–364
Hayafunea M, Berisiob R, Marchettic R, Silipoc A, Kayamaa M, Desakia Y, Arimaa S, Squegliab F, Ruggierob A, Tokuyasud K, Molinaroc A, Kakua H, Shibuyaa N (2014) Chitin-induced activation of immune signaling by the rice receptor CEBiP relies on a unique sandwich-type dimerization. Proc Natl Acad Sci 10:1073
Jiang C, Wen Q, Chen Y, Xu L, Huang M (2011) Efficient extraction of RNA from various Camellia species rich in secondary metabolites for deep transcriptome sequencing and gene expression analysis. Afr J Biotechnol 10:16769–16773
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444:323–329
Lee SJ, Rose JKC (2010) Mediation of the transition from biotrophy to necrotrophy in hemibiotrophic plant pathogens by secreted effector proteins. Plant Signal Behav 5:769–772
Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, Hammond-Kosack KE, Thomma BP, Rudd JJ (2011) Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol 156:756–769
Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, Saitoh H, Terauchi R, Nishizawa Y, Shibuya N, Thomma BP, Talbot NJ (2012) Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. The Plant Cell 24:322–335
Mitsuhara I, Matsufuru H, Ohshima M, Kaku H, Nakajima Y, Murai N, Natori S, Ohashi Y (2000) Induced expression of sarcotoxin IA enhanced host resistance against both bacterial and fungal pathogens in transgenic tobacco. Mol Plant-Microbe In 13:860–868
Morris K, Thornber S, Codrai L, Richardson C, Craig A, Sadanandom A, Thomas B, Jackson S (2010) DAY NEUTRAL FLOWERING represses CONSTANS to prevent Arabidopsis flowering early in short days. The Plant Cell 22:1118–1128
Shimizu T, Nakano T, Takamizawa D, Desaki Y, Ishii-Minami N, Nishizawa Y, Minami E, Okada K, Yamane H, Kaku H, Shibuya N (2010) Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J 64:204–214
Spiers AG (1983) Host range and pathogenicity studies of Marsonnina brunnea to poplars. Eur J For Pathol 13:181–196
Wu R, Wang M, Huang M (1992) Quantitative genetics of yield breeding for Populus short rotation culture. I. Dynamics of genetic control and selection model of yield traits. Can J For Res 22:175–182
Xu M, Zhang B, Yao H, Huang M (2009) Isolation of high quality RNA and molecular manipulations with various tissues of Populus. Rus J Plant Physiol 56:716–719
Zhu S, Cao Y, Jiang C, Tan B, Wang Z, Feng S, Zhang L, Su X, Brejova B, Vinar T, Xu M, Wang M, Zhang S, Huang M, Wu R, Zhou Y (2012) Sequencing the genome of Marssonina brunnea reveals fungus-poplar co-evolution. BMC Genomics 13:382
Acknowledgements
This research was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Collaborative Innovation Plan of Jiangsu Higher Education. We acknowledge Professor Dacheng Tian and Yonghua Yang from Nanjing University who provide us A. thaliana Col-0.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
PCR primers used in the real-time PCR assay. (DOCX 14 kb)
Fig. S1
Sequence analysis of LysM genes and LysM domains. (A). Multiple sequence alignment of LysM genes in Marssonina.brunnea; (B). Consensus sequence of LysM domain (GIF 432 kb)
Rights and permissions
About this article

Cite this article
Jiang, C., He, B., Huang, R. et al. Identification and functional analysis of LysM effectors from Marssonina brunnea . Australasian Plant Pathol. 43, 615–622 (2014). https://doi.org/10.1007/s13313-014-0316-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13313-014-0316-5