
Abstract
Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) are termed synucleinopathies, disorders that are characterized by the intracellular aggregation of the protein ɑ-synuclein. The cellular tropism of synuclein pathology in these syndromes is notably distinct since in the Lewy disorders, PD and DLB, ɑSyn forms aggregates in neurons whereas in MSA ɑSyn forms aggregates in oligodendrocytes. Studies examining ɑSyn pathology in experimental models and in human brain have now identified fibrillar ɑSyn with unique but distinct molecular signatures, suggesting that the structure of these ɑSyn fibrils might be closely tied to their cellular ontogeny. In contrast to the native structural heterogeneity of ɑSyn in vitro, the conformational landscape of fibrillar ɑSyn in human brain and in vivo transmission models appears to be remarkably uniform. Here, we review the studies by which we propose a hypothesis that the cellular host environment might be in part responsible for how ɑSyn filaments assemble into phenotype-specific strains. We postulate that the maturation of ɑSyn strains develops as a function of their in vivo transmission routes and cell-specific risk factors. The impact of the cellular environment on the structural diversity of ɑSyn might have important implications for the design of preclinical studies and their use for the development of ɑSyn-based biomarkers and therapeutic strategies. By combining phenotype-specific fibrils and relevant synucleinopathy transmission models, preclinical models might more closely reflect unique disease phenotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The synucleinopathies are a group of degenerative brain disorders that are characterized by intracellular deposits of the protein ⍺-synuclein (ɑSyn). Inclusions of aggregated ɑSyn are the pathognomonic feature of synucleinopathies as they are invariably found in affected areas of the peripheral and central nervous system. Synucleinopathies include Parkinson’s disease (PD), dementia with Lewy bodies, multiple system atrophy (MSA), and other related but less common disorders, such as pure autonomic failure. Genetic variants as well gene duplications and triplication of ɑSyn give rise to early onset Parkinson’s disease directly linking ɑSyn to familial and sporadic cases of PD [1, 2]. PD and Lewy body dementia are characterized by cell-specific deposits of ɑSyn in neurons [3], termed Lewy body inclusions, whereas in MSA ɑSyn aggregates are observed in oligodendrocytes named glial cytoplasmic inclusions [4]. Because of the neuronal tropism of synucleinopathy in PD and DLB, these disorders are collectively referred to as Lewy diseases.
The main constituent of Lewy bodies or glial cytoplasmic inclusions is fibrillar ɑSyn. These filamentous forms of ɑSyn have been extracted from postmortem brain but can also be isolated from cerebrospinal fluid of patients with Lewy diseases or MSA [5,6,7,8]. Purified ɑSyn filaments from the brain of people with the same clinical diagnosis show a remarkable overlap in their conformation [5,6,7]. Contrastingly, when comparing Lewy disorders and MSA, the conformation of the ɑSyn fibrils is noticeably distinct. Fibrils of ɑSyn thus have disease-specific conformations, reflecting a molecular fingerprint that is intimately linked to clinical diagnosis [5,6,7]. Diagnosis of PD, DLB, or MSA is currently based on clinical consensus criteria but because of the conformational uniformity of ɑSyn filaments in Lewy diseases and MSA, ɑSyn filaments are being investigated as a proximal diagnostic marker via seeded amplification assays [9, 10].
Many studies have shown that fibrillar ɑSyn is intrinsically pathogenic [11,12,13], and that ɑSyn aggregates can cause cellular dysfunction resulting in white and gray matter degeneration in experimental models. Fibrillar aggregates of ɑSyn can be transmitted between cells in vitro and in vivo [14, 15]. Stable filamentous seeds of ɑSyn can replicate intracellularly with endogenous ɑSyn and form new ɑSyn fibrils in a manner that resembles infectious proteins. Because of the compelling evidence for the pathobiology of fibrillar ɑSyn in the neurodegenerative process, fibrillar assemblies of ɑSyn are now widely used for animal modeling or preclinical studies that inform clinical trials [11].
However, there are still several challenges in translating ɑSyn-related pathology from preclinical models to humans. First, we need to better understand the molecular basis of the clinical diversity in synucleinopathies, as it has been difficult to reconcile the observation of ɑSyn as a key pathogenic substrate with different pathologies across a spectrum of related human syndromes. Adding to this complexity is the species barrier between rodent and human, considering that the structural features of human ɑSyn have mostly been studied after intracellular replication in rodent models. More accurately capturing the structural diversity of human ɑSyn assemblies within this experimental setting would benefit the development of future preclinical trials and allow to make better predictions for ɑSyn-targeting therapeutic strategies in humans.
Another not well understood key factor in ɑSyn transmission is the intracellular or in vivo environment. With recent methodological developments, the role of the host environment in ɑSyn pathobiology becomes increasingly appreciated as the cellular environment appears to be closely tied to the structural identity of its populating conformers [16, 17]. Next to characterizing the structural properties of ɑSyn aggregates, understanding how aggregation is initiated and proceeds within the cellular host environment might be crucial to understand the similarities and the differences in the development of synucleinopathies. Preclinical animal models would thus need to mimic and establish the conditions wherein the folding landscape of pathogenic ɑSyn resembles that of the human brain—on the cellular and the structural level. Here, we will discuss emerging insights into the native function and structural diversity of ɑSyn and the transmission of pathology in the context of the host cellular environment in preclinical ɑSyn transmission models. We further highlight the advantages and shortcomings of transmission models and propose how to address outstanding research gaps.
Native and Aggregated Assemblies—The Conformational Diversity of ⍺-Synuclein
The relationship between the conformation and the function of ɑSyn has been intensively studied for more than two decades, but its native role remains elusive partly because of structural heterogeneity of the protein. There are significant methodological challenges to study ɑSyn as it is a small intrinsically disordered protein, with an apparent lack of conformation. It is expressed widespread in various tissues of the body, in- and outside the brain, underscoring its functional diversity. It is highly abundant in the human central and the peripheral nervous system [18, 19] and in rodent brain it is one of the most abundant synaptic proteins [20]. The expression of ɑSyn in the peripheral nervous system is also evident at barrier sites, such as the skin or visceral organs. Often, deposits of ɑSyn are observed within these barrier sites but in the absence of clear signs of cellular pathology or neurological illness [21, 22]. These peripheral deposits could also be tied to the presence of ɑSyn in the blood, as ɑSyn is also present in leukocytes and erythrocytes, where it is highly enriched [23].
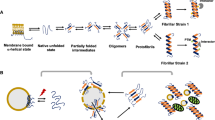
ɑSyn is a small protein of 140 amino acids which lacks a defined structure in solution. When incubating the protein with negatively charged membranes, the N-terminus of ɑSyn adopts an ɑ-helical structure similar to apolipoproteins via inserting its positively charged and hydrophobic KTKEGV repeats (Fig. 1) into the phospholipid bilayer [13]. This is followed by binding of the central core of the protein via its more hydrophobic non-amyloid component (NAC) region with the membrane [24, 25]. The carboxy terminus of protein remains unstructured as it is highly negatively charged and does not bind with the membrane [26, 27].

Schematic of ɑSyn native and pathological conformations. a ɑSyn is a small protein of 140 amino acids with an N-terminal amphipathic region, a central hydrophobic region, and a highly charged acidic tail. Interactions with phospholipid membranes are mediated via consensus KTKEGV repeats in the N-terminus and the NAC region. b Monomeric ɑSyn is unstructured and natively unfolded in solution. Under pathological conditions, ɑSyn monomers will form unwanted intermolecular interactions via β-sheets through its polypeptide backbone. Upon assembly into stable oligomeric species, larger ɑSyn filaments can form, which are deposited into Lewy bodies and which are characteristic of synucleinopathies
In neurons of the peripheral and central nervous system, ɑSyn is predominantly located in the neuronal synapse where it preferentially binds with the outer membrane of synaptic vesicles to mediate synaptic vesicle endo- and exocytosis [28]. ɑSyn is also present in the cytoplasm as it interacts with membranes of high curvature such as mitochondria, granules, or other smaller organelles [29, 30]. More recently, it was shown that ɑSyn binds with cytosolic mRNA proteins on membraneless organelles via its N-terminus [31]. By doing so, it stabilizes translationally repressed mRNA transcripts in granular processing bodies, slowing down their degradation [31].
As an intrinsically disordered protein with no discernable conformation, ɑSyn can undergo conformational changes when it inserts itself into the outer lipid bilayer of curvature-rich membranes. Upon binding with the membrane, the protein transitions to a membrane bound helical state via which it stabilizes lipid packaging by relieving the elastic membrane stress on the outer membrane of the membrane vesicle [32]. On these small vesicles, membrane-bound ɑSyn can transiently interact with other ɑSyn monomers, to form helical multimers on the membrane [33]. These structural changes of ɑSyn occur fast, and thus strongly rely on the conformational flexibility of the protein [34, 35].
Although this has been frequently overlooked, next to its ubiquitous expression in the nervous system, ɑSyn is highly abundant in the blood [23], the epithelial vasculature [36, 37], and in cells of the innate immune system such as granulocytes and other white blood cells [38, 39]. The transcriptional expression levels of ɑSyn in some human leucocytes, such as dendritic cells, are comparable to the levels of expression in other cell types of the brain [39, 40]. ɑSyn expression is low in cells of the adaptive immune system, such as in lymphocytes [39, 40]. In different types of innate leukocytes, ɑSyn is enriched on vesicular and granular membranes. Because its membrane binding role and granule stabilizing capacities, it likely plays a role in granule vesicle dynamics in leukocytes [41]. However, it is still unclear what its role is of ɑSyn in leucocytes and especially erythrocytes, where ɑSyn is highly abundant.
Recent studies have shown that ɑSyn is important for the host–pathogen immune response [42] as it plays a critical role in innate immune defense mechanisms [43,44,45]. Next to its putative role in the peripheral immune system, ɑSyn function is also critical to mediate the central immune response. Expression of ɑSyn in the brain during infection has been shown vital for protection against acute viral encephalitis as mice without ɑSyn lack a host pathogen response and succumb to infection [44,45,46]. Neurotropic infection triggers neuronal ɑSyn expression in the brain. This causes the protein to transiently accumulate in the neuronal cytoplasm and redistribute intracellularly as it relocates into the cell nucleus to supports the expression of interferon-related genes [44, 47].
ɑSyn is thus expressed in a variety of cells in and outside the brain. The structural diversity or structural flexibility of ɑSyn is imperative for its function as the protein has a broad impact on a wide variety of cellular functions. With its cellular distribution in the cytosol and membranes, ɑSyn needs to efficiently transition between different subcellular compartments to fulfill its native function. The conformational flexibility is therefore a functional advantage since it allows ɑSyn to rapidly perform complex dynamic interactions. However, unwanted interactions between ɑSyn monomers can as quickly turn this advantage into a disadvantage. Folding intermediates of ɑSyn on the membrane or in the cytosol can interact because of exposed hydrophobic residues in the NAC region, which are normally shielded by charged residues of the protein itself. However, exposure of these residues allows multiple molecules to bind and form high molecular weight assemblies. These aggregates of ɑSyn can set off a pathogenic cascade of protein aggregation with potentially far-reaching complications.
Although the structural flexibility of ɑSyn serves an important functional role, it can thus lead to unwanted interactions between multiple ɑSyn molecules. Because of the dynamic interactions of ɑSyn monomers with its environment, the protein can expose hydrophobic sequences via an intermediate assembly state that now allows direct interactions with other ɑSyn monomers [48, 49]. The polypeptide backbone of two or more ɑSyn molecules can transiently bind and form β-sheets leading to their aggregation into small but unstable oligomers [50, 51]. These amorphic oligomers need to undergo structural reorganization before they become stable and assemble into an aggregate core that forms the basis of a protofilament [48,49,50,51,52,53]. The filament can now serve as a seeding nucleus, with which new ɑSyn monomers can nucleate at the fibrillar surface catalyzing the formation of new aggregates or with which monomers can bind at the ends of the filament resulting in fibrillar elongation [54].
Oligomers and stable filaments of ɑSyn can exist in a range of sizes of shapes. There have been extensive efforts to characterize the functional or pathological effects of different types of ɑSyn conformers. The smaller variants of ɑSyn oligomers are particularly challenging to study due to their kinetically unstable nature, their vast conformational heterogeneity, or subgroups of oligomeric assembly states. These oligomers can have distinct structures with various extents of β-sheet content or hydrophobicity.
Nevertheless, many studies have found that several of the smaller oligomeric assemblies can have detrimental effects on a wide range of cellular biological processes in vitro [50, 55]. Structural conversion of newly formed oligomers into more stable assemblies often leads to the formation of assemblies with more lasting and therefore more damaging effects on cellular health [53, 56, 57]. Even upon transient rearrangement into intermediate folding states in vitro, there can be high kinetic barriers during structural conversion [53]. More structurally defined oligomers can insert into the membrane bilayer via binding of amphipathic helices to the outer membrane and inserting their β-sheet into the deeper layers, thereby disrupting membrane integrity [58].
The existence of distinct subgroups of oligomers is likely due to the multiplicity of pathways in the misfolding process although the exact determinants required for the structural assembly into unique small oligomers are not yet fully understood. However, assembly conditions during in vitro aggregation can significantly influence how oligomeric or filamentous aggregates assemble, with slight variations in buffer composition or shaking conditions yielding different structural outcomes in the assembly state of the aggregate [48, 59]. Indeed it was shown that different in vitro assembly conditions via varying the composition of salts in the aggregation buffer led to the assembly of two types of fibrils with distinct structures as defined by the fibrillar morphological, biophysical, and biochemical properties [60]. The resulting assemblies were termed fibrils and ribbons, in accordance with their morphological appearance. In cell culture, the two types of fibrils amplified with endogenous ɑSyn but they exhibited distinct cellular toxicities [60]. There is thus a structural functional relationship between ɑSyn assemblies and it cellular phenotypes. Because of this direct relationship between aggregate structure and function, it is imperative that studies investigating the biological effects of ɑSyn assemblies are well controlled, by clearly describing the purification and assembly conditions and by making efforts to work with structurally related or homogeneous assemblies instead of heterogeneous mixtures, to infer reliable and consistent results within and between studies.
Fibrillar Assemblies—The Conformational Uniformity of ⍺-Synuclein Strains
Contrasting with the heterogeneity of structural assemblies formed in vitro is the structural homogeneity of ɑSyn filaments formed in vivo. Using cryo-electron microscopy, recent studies have shown that fibrils postmortem extracted from the brain of six unrelated people with Lewy disease contained only a single filament with strikingly matching conformations between isolated filaments [5, 7]. These filaments had a typical fold, seen in the outer and inner layers of the fibril core, and it very closely matched the structure of the fibrils isolated from the brain of the people with either PD, DLB, or PD with dementia [5, 7]. Even though ultrastructural differences were still noticeable in the fibrillar rotation between the filaments analyzed from people with a clinical diagnosis of PD and DLB, the structure or the fold within the fibrillar core of the ɑSyn fibrils was almost indistinguishable. To indicate the structural resemblance of this unique fold from fibrils isolated from patients with Lewy disorders, this conformation was termed a “Lewy fold” [5, 7].
In MSA, different assembly conditions likely exist, as the cellular tropism of synucleinopathy is mostly directed towards oligodendrocytes instead of neurons and in which the fibrillar assembly of ɑSyn thus might be differentially influenced. Postmortem structural analysis of fibrils derived from the brain of three people with MSA has shown that ɑSyn fibrils are composed of two filaments that interact via their outer surface [5]. The outer filament layers pack and stabilize the conformation of the resulting fibril via a shared interface in which an unidentified substrate screens charged residues in the fibril cavity that allow the two filaments to closely interact [5]. The two filaments slightly differ in their conformation so that the fibril is asymmetrically composed by two non-identical filaments that twist around a central axis [5].
Although the fibrils from PD and DLB (or PD with dementia) show slight differences in their overall structure, the core of the two types of PD and DLB fibrils is surprisingly similar. They both have a typical Lewy fold but with a slightly different pitch, in which the fibrils turn around their own axis [5, 7]. Although these results remain to be replicated in larger studies, the similarities between fibrils of Lewy disorders tentatively suggest that these fibrils have shared ontologies and that Lewy disorders could be part of an overlapping or continuous disease spectrum where cellular conditions are present under which structurally similar proteopathic seeds can assemble in neurons. This largely contrasts with the disease conditions in MSA, where oligodendrogliopathy or oligodendroglial cellular risk factors might influence the formation of a different type of fibril [16, 17].
Because of the unique folds in the conformation of the fibrils from Lewy diseases and MSA brain, the pathogenic blueprint of distal pathology might be imprinted within the conformation of these fibrillar strains. The cellular interactome and the conditions in which fibrils assemble can have a strong impact on the amplification and the structure of the resulting fibril [6]. Nevertheless, establishing the cellular conditions in which ɑSyn strains form and why they develop their typical structural features remains largely unexplored. To understand the etiopathogenesis of synucleinopathies, it will be important to further address these questions.
Fibrillar Transmission in Animal Models of Synucleinopathy
An important aspect of fibrillar in vivo toxicity is its sustained or infectious pathogenicity via filaments that seed with endogenous monomers to form mature inclusions, thereby disrupting central cellular processes [61]. Initial evidence that recombinant ɑSyn fibrils could cause pathology in vivo came after injecting fibrillar ɑSyn in transgenic mice (M83) bearing the familial A53T mutation of ɑSyn [62]. Inclusions of ɑSyn phosphorylated on serine at position 129 (pSer129-ɑSyn) in the striatum, substantia nigra, and connected areas progressively worsened over time. This accumulation of pSer129-ɑSyn was accompanied by moderate dopaminergic degeneration and behavioral impairment [62]. Following this, a study by the same group showed that striatal injection of fibrillar ɑSyn could also trigger de novo formation of pSer129-ɑSyn pathology in wild type mice (C57BL6). The authors again observed a spatiotemporal pattern of pSer129-ɑSyn pathology from the site of injection with neurodegeneration [63]. These findings were important in the light of the transmission hypothesis and provided the first evidence that ɑSyn fibrils can seed pathology in vivo.
In support of this view, subsequent studies independently confirmed that injection of in vitro assembled ɑSyn fibrils in wild type mice and rats can seed in vivo ɑSyn aggregation [64,65,66,67]. Direct live imaging of ɑSyn inclusion formation in mouse cortex showed that cortical neurons selectively degenerate during inclusion formation [68]. This is in accordance with other studies that established a close relationship between ɑSyn inclusion formation and cellular toxicity during seeding [61]. Injection of fibrillar ɑSyn and not oligomeric ɑSyn seems to be crucial for inclusion formation [69]. Although oligomeric ɑSyn can cause local neuronal damage, only stable fibrillar seeds can trigger the conversions of endogenous ɑSyn into new aggregates akin to Lewy pathology [69, 70]. Small fibrillar aggregates (in the range of 50–100 nm) are potent seeders and more efficiently cause inclusions and pathology than their larger fibrillar counterparts [71].
However, cellular degeneration also appears to be cell-specific and selectively affect vulnerable populations of cells over time [72, 73]. Some neurons might therefore remain resilient and show no degenerative changes, even after fibrillar uptake. This cell-type specific vulnerability and resilience is an intricate mechanism of ɑSyn cell-to-cell transmission and will be further discussed in one of the following sections. Altogether, ɑSyn transmission models using in vitro generated fibrils have now become invaluable for designing and testing new strategies that intervene with ɑSyn-related mechanisms of neurodegeneration.
Strain-Specific Transmission in Animal Models of Synucleinopathy
Given the cellular tropism of pathology in synucleinopathies and the neuropathological changes seen in experimental models during ɑSyn transmission, research has focused on whether phenotype-specific effects could be transferred by the fibril conformation. As it was shown by in vitro intracellular seeding, different strains of ɑSyn, ribbons and fibrils, amplified into distinct types of inclusions that conferred phenotype-specific effects [60]. These two type of strains were subsequently injected into the substantia nigra of rats [70]. Both fibrils amplified in vivo and caused phenotype-specific effects while retaining their conformation-specific properties [70]. Because of the relationship between structure of ribbons and fibrils and the cellular phenotypes that were inseparable from the two fibrillar types, the two in vitro generated fibrils thus behaved as protein strains.
The relationship between the structural diversity of ɑSyn strains and developing in vivo phenotypes was further shown via other approaches. This involved the generation of ɑSyn fibrils via altering the pH or other salts in the buffer composition, via repetitive seeding or via adding cell-specific or environmental cofactors relevant to PD or MSA that might influence the folding of ɑSyn [70, 74,75,76,77,78]. As such, repetitively in vitro seeded ɑSyn fibrils caused tau pathology in vivo whereas first generation or de novo assembled fibrils did not [58]. Incubation of ɑSyn with the bacterial endotoxin lipopolysaccharide, or the oligodendroglial protein p25, resulted in the assembly of unique fibrils that amplified in vivo with strain-specific toxicities. The conformational variation of distinct fibrils also largely determined which cellular populations were targeted and the brain regions that developed pathology [76, 77]. These phenotypes were furthermore retained after serial passaging between different animals emphasizing a mechanism that involves conserved templating-directed amplification of the injected strain in vivo [77]. Collectively these studies thus show that recombinant fibrils of ɑSyn with a defined conformation can act as strains, providing strong evidence for a structural-pathological relationship between the conformation of the ɑSyn fibril and the developing phenotype [58].
Next to using recombinant assembled fibrils, strain-related pathologies have also been studied by using brain extracts or by purifying and amplifying ɑSyn fibrils from patient brain for in vivo transmission (Fig. 2). In the case of brain extracts, pathological brain is homogenized and directly injected intracerebrally in wild type or transgenic animals [64, 79,80,81,82,83,84,85,86]. Several studies have compared intracerebral injection of brain homogenates from people with PD, DLB, or MSA with similar findings: MSA brain homogenates are generally more toxic and cause more aggressive neurodegeneration with neuroinflammation than brain homogenates derived from people with Lewy disorders [64, 81,82,83,84,85].

Seeded amplification of ɑSyn from human brain. a For the amplification of ɑSyn from human brain, brain tissue is isolated from selected brain regions and homogenized. Samples are sonicated to fractionate ɑSyn fibrils into smaller fibrillar seeds. b Monomeric ɑSyn is added with amplification buffer and in vitro templated amplification takes place under shaking conditions using the fractionated seeds from the brain homogenate. Fibrils will elongate after incorporation of new monomers at the fibril ends via templating-directed amplification and building on the original input conformation. After each reaction, amplified samples are diluted and sonicated to generate new fibrillar fragments. c When this process is repeated several times, highly homogenous fibrillar assemblies, free from brain material, will be available for subsequent experimental work in animal models
In line with these observations, experiments with fibrils purified and amplified from human brain have shown similar outcomes [81, 87]. Here, after isolating ɑSyn fibrils from patient brain, fibrils are amplified via seeded amplification and the residual brain material after extraction is diluted to levels at which it becomes undetectable (Fig. 2). By controlling the in vitro assembly conditions of ɑSyn, seeded amplification will generate ɑSyn fibrils that are directly derived from human fibrillar seeds. After serial sonication and amplification by shaking and incubation with recombinant monomers, the structural information from brain-derived seeds is at least partially templated onto the newly assembled fibrils [6, 88] (Fig. 2). After injection of fibrils derived from PD, DLB, and MSA brain in rodent brain, animals develop distinct phenotypes, depending on the type of fibril [81]. Reminiscent of experiments with brain homogenates, fibrils derived from MSA brain induced the most progressive phenotype, with more pronounced PSer129-ɑSyn inclusions around and away from the injected site [81]. In human induced pluripotent stem cells (iPSC) derived dopaminergic neurons, the aggregate burden of MSA or PD fibrils was determined by ɑSyn expression levels and the type of strain administered to the cells [89]. Fibrils derived from the brain of people with distinct clinical syndromes thus uniquely affect cellular health and transmit pathology to distinct cell types or brain areas, again illustrating that ɑSyn strains and the cellular environment strongly interact.
Both materials, brain homogenates or seeded and templated aggregates, have advantages as well as disadvantages for preclinical animal studies as injection of brain homogenates will contain unmodified ɑSyn aggregates with their post-translational modifications whereas amplified fibrils do not have the same modifications. Fibrils isolated from Lewy bodies have several post-translational modifications, including phosphorylation, acetylation, ubiquitinylation, C-terminal truncations, among many others [90,91,92,93]. Notable differences are also found in the distribution of these modifications in ɑSyn isolated from brains with either Lewy or MSA pathology [93]. Modified soluble ɑSyn, such as PSer129-ɑSyn, can influence the seeding, transmission, and pathogenicity of ɑSyn fibrils in vitro and in vivo [93,94,95]. In contrast, the protein misfolding cyclic amplification method amplifies fibrils with recombinant ɑSyn that is largely free of post-translational modifications; however, using this method, it more strictly avoids inoculating any potential host-specific pathological triggers, present in brain homogenate, such as inflammatory mediators, that could set off unwanted ɑSyn aggregation. Altogether, studies using either brain homogenates or brain-derived amplified seeds provide additional support that ɑSyn strains can be amplified from human brain and that unique structural information can be transferred via seeded amplification. This structural information can also transmit disease-specific pathology in vivo.
The Cellular Milieu Impacts Fibrillar Transmission—Lewy Disorders
Significant evidence thus supports a role for ɑSyn strains in the development of synucleinopathies and ɑSyn fibrils from antemortem and postmortem samples have a unique disease-specific fold that is intimately tied to a clinical diagnosis of Lewy disease or MSA [5,6,7,8]. But why is the fold of fibrils between different syndromes so different—or, how do ɑSyn fibrils obtain their unique conformation?
The structural and cellular heterogeneity of native ɑSyn is astounding, and the potential conformations that ɑSyn can adapt are almost limitless. Within these lines, multiple studies have shown that by varying aggregation conditions, numerous structural variations in the fibrillar conformation can be obtained [48, 59, 60, 70, 74,75,76,77]. Nevertheless, there seems to be a remarkable uniformity between the conformation of ⍺Syn fibrils that are isolated from unrelated patients that share a similar clinical diagnosis. The solved structures of fibrils from patients with Lewy diseases—albeit a limited group—are almost indistinguishable, and although the fibrils from MSA patients do show subtle differences, depending on the region from where the fibrils were isolated, their quaternary organization and filament outer and inner core layers are closely similar [5]. Together with the experimental evidence that patient-derived fibrils of ɑSyn can cause disease-specific phenotypes in experimental models, it can be concluded that the conformational landscape of aggregated ɑSyn in patient brain is relatively narrow, which is counterintuitive from the many possible conformations that ɑSyn fibrils could potentially and experimentally adapt. This now leaves the question of how these disease-specific assemblies arise or why only a restricted population of fibrillar strains exists within a patient’s brain.
Although addressing this question can be experimentally challenging, cellular and in vivo models have provided important clues as to whether cell autonomous factors might limit or promote the transmission of pathology. Concurrently, these cellular factors could be equally responsible for the maturation of ɑSyn into phenotype-specific fibrils. On the level of cellular connectome, there are several determinants that could influence the transmission of ɑSyn fibrils. First is the susceptibility or resilience of the incipient or recipient cell in which fibrils assemble or transmit (the cell type). Secondly, transmission is strongly dependent on ɑSyn levels with which fibrils can amplify (ɑSyn expression). A last factor is the strength of projections between the cells in which fibrils amplify and to which the seeds are transmitted (the connectome).
In normal human brain, as well as in the brain of people with PD and MSA, the highest levels of SNCA gene expression is in neurons, followed by microglia and mature oligodendrocytes, where expression is still significant but lower [19, 40]. Although directly comparing ɑSyn expression between human and mouse brain has not yet been possible, a similar distribution is seen in mouse brain [18]. Several studies have examined the pattern of transmission in mouse brain, by injecting fibrillar ɑSyn in defined brain areas. Based on the observation of pSer129-ɑSyn inclusions, the transmission of pathology was shown to be dependent on the levels of endogenous ɑSyn but also on the cell type in which pathology develops [96,97,98]. As such, these experimental models show that seeded aggregation as well as transmission of ɑSyn is a function of brain connectivity and ɑSyn protein levels.
Notably, pSer129-ɑSyn-specific pathology can be transient, as regions that develop pSer129-ɑSyn inclusions have been shown to eventually clear pSer129-ɑSyn pathology [72], indicating that in addition to the neuronal connectome and cell-autonomous factors, there can be neuronal resilience that counteracts in vivo transmission. Contrarily, genetic or environmental risk factors can facilitate ɑSyn fibrillar permissivity. For instance, GBA or LRRK2 genetic variants can unlock a disease-associated cellular environment in which the barriers for fibrillar transmission are lowered [99,100,101,102], although the exact mechanisms behind this are not yet clear [103]. The propagation of ɑSyn appears thus restricted in certain subsets of cells but facilitated by others. These features thereby define the cellular subtype as a last determinant of fibrillar transmission, in which fibrils can either become degraded or amplify in conjunction with PD-associated host risk factors.
In light of the strain hypothesis, the serial transmission through a permissive neuronal route could thus be a conditio sine qua non for the assembly of conformation-specific filaments as it would almost invariably lead to a defined path down a folding landscape in which only a restricted set of fibrillar assemblies with high thermodynamic stability can exist. As the host cellular environment both restricts as well as facilitates the formation of fibrillar strains, a predominant filament can further amplify within its disease-associated cellular environment on the background of the aforementioned neuronal determinants.
What further illustrates this idea is that although strains propagate phenotype-specific effects in vivo, in vivo amplification within a defined cellular environment can lead to measurable conformational changes in the folding landscape (Box 1). By directly measuring the conformational spectrum using brain-penetrant conformation-sensitive fluorescent probes, it was shown that the conformation of pSer129-positive ɑSyn aggregates in Lewy bodies is strongly restricted, with a low degree of conformational variability [104]. Similarly, the folding landscape of pSer129-positive ɑSyn aggregates in oligodendrocytes is narrowly distributed, but shows a conformational spectrum that does not overlap with that of Lewy bodies [17, 104]. The use of these oligothiophene probes does not require any extraction or fibril amplification and they measure the structural state of aggregates in their respective environments in situ. These results therefore corroborate that ɑSyn fibrils in neurons and oligodendrocytes adopt distinct conformations within their respective cellular environment.
Other studies that examined the composition of Lewy bodies in situ have also found remarkable findings. Lewy bodies are complex intracellular deposits composed of complex lipid structures and multiple ɑSyn assemblies with distinct PTMs, depending on its distribution within the Lewy body [91, 92]. The initial steps of ɑSyn aggregation might take place on the membrane, and more in particular on membranes that have a particular affinity for ɑSyn, thereby acting as an aggregation hot spot, as is for instance seen for mitochondria, the nuclear membrane, or the Golgi membrane [61, 105]. There is thus a strong enrichment in Lewy bodies of proteins and undigested membranes, from dystrophic mitochondria, lysosomes, or other lipid-rich damaged organelles in between which ɑSyn fibrils are sequestered [90,91,92, 106, 107].
The cellular proteome and lipidome could thereby act as a crucial cofactor for templated seeding of unique ɑSyn assemblies into fibrillar inclusions or ɑSyn strains. Future studies will have to identify the cellular identity of the transmissible connectome and the cellular cofactors that conduct the amplification of heterogeneous assemblies into phenotype-specific strains. Reconstructing the protein and lipid interactome as well as the connectome of sequentially transmitted ɑSyn fibrils in humanized models could provide important new clues as to which cofactors and cellular risk factors might govern these processes. Identifying the molecular and cellular identity of a strain-permissive environment could be very valuable as this could inform the development of novel in vitro or in cellulo seeding assays for assembling phenotype-specific recombinant PD, DLB, or MSA ɑSyn strains, as it has been successfully done for other amyloid proteins.

The Cellular Milieu Impacts Fibrillar Transmission—MSA
In MSA, pathological changes are accompanied by a high burden of oligodendroglial ɑSyn in areas with dominant oligodendropathy and neuronal degeneration [109, 110]. Although it is not yet known how MSA arises, several experimental findings point to a strong link between ɑSyn aggregation, oligodendropathy, and the degenerative process. Experiments with various animal models of MSA that selectively overexpress human ɑSyn in oligodendrocytes have shown that ɑSyn overexpression is sufficient to cause oligodendropathy, resulting in structural and functional in vivo changes that in part resemble those of MSA (reviewed in [11]). Intracerebral injection of MSA brain homogenates or amplified fibrils from MSA brain causes neurodegeneration in wild type and transgenic animal models [79, 81, 82, 84, 85]. When compared to ɑSyn assemblies derived from the brain of people with Lewy disorders, MSA-derived fibrils are invariably more pathogenic.
The more aggressive nature of MSA fibrils might explain why MSA is a more progressive disease than Lewy disorders, but some crucial aspects as to how the typical pathology of MSA is caused remain puzzling. For example, direct intracerebral injection of MSA fibrils in animal models causes primarily neuronal pathology but has mostly failed to trigger oligodendroglial dysfunction. There is thus still a discrepancy between the strain-related phenotypic effects and the development of oligodendropathy in experimental models of MSA.
As such, there might be a unique but elusive role of the oligodendroglial cellular environment in the development of MSA. Although the insoluble proteome in human PD and MSA brain shows a significant overlap in mitochondrial and neuronal synaptic proteins [111], the potential interactome of ɑSyn in oligodendrocytes could be very different from that in neurons as both the proteome and especially the lipidome in oligodendrocytes are unique [112, 113]. The role of ɑSyn in oligodendrocytes remains largely unexplored and it has been questioned if oligodendrocytes can express sufficient endogenous ɑSyn to form intracellular aggregates [114, 115]. Nevertheless, despite some contradictory reports, expression of ɑSyn is detected in oligodendrocytes of the human and rodent brain [18, 19, 116, 117], albeit at considerably lower levels compared to neurons. Some disease-associated subtypes of oligodendrocytes in human brain have also been shown to express higher levels of ɑSyn [118]. Experimental findings point towards an active role of oligodendrocytes that involves template-directed amplification, and which would require the expression of ɑSyn in oligodendrocytes [16, 17]. It has been shown that by passaging fibrillar ɑSyn in the oligodendrocytes, a unique type of fibril develops as a consequence of repeated passaging within the oligodendroglial milieu [16, 17]. The conformation of MSA filaments is unique as they have a distinct fold with two filaments positioned around a polar cavity between the two central layers, which are screened by an unidentified residue [5]. This residue allows the two protofilaments to come closely together and interact. Why MSA fibrils have this unique feature is not yet known, but it likely happens via yet unidentified oligodendroglial factors that facilitate the templated seeding of MSA-specific fibrils.
The oligodendroglial cellular environment can thus introduce significant structural variations in the conformational state of the fibril and as a result an oligodendroglial-specific strain will develop [16]. Oligodendroglial strains amplify faithfully in oligodendrocytes, and interestingly, the same conformation can be transmitted to neurons, which apparently cannot convert the new conformation back to a neuronal strain [16]. Although this remains to be experimentally tested, it raises the possibility that MSA strains can be transmitted between oligodendrocytes and neurons, and that the predominant strain, formed in oligodendrocytes, could be passaged between neurons and oligodendrocytes while retaining most of its oligodendroglial structural features and its aggressive cellular pathogenicity. This would eventually lead to a conformational landscape with a restricted number of fibrillar variants in MSA brain [5, 17, 104, 119], as its conformation would be largely determined by host-specific cellular factors.
Furthermore, the interactions during templated amplification in oligodendrocytes seem to be bidirectional, as interactions also exist between the fibrillar strain and the intracellular milieu. Indeed, intrastriatal injection of two different types of ɑSyn strains, ribbons and fibrils, in MSA transgenic mice overexpressing oligodendroglial ɑSyn revealed that the ɑSyn conformational landscape not only depends on cell autonomous factors, but also on the transmitted strain [17]. For instance, compared to fibrils, injections of ribbons more closely represented the conformational profile of glial cytoplasmic inclusions of the human brain [17]. This was accompanied by a narrowing of the conformational landscape, indicating that a predominant form arose after amplification in vivo (Box 1 and [17]). Thus, combining the selective overexpression of oligodendroglial ɑSyn with intracerebral injection of ɑSyn fibrils resulted in a phenotype that more closely mimics MSA [16, 17]. Together, it can be postulated that for Lewy disorders as well as for MSA ɑSyn aggregation can occur actively in either neurons or oligodendrocytes via templated seeding with endogenous ɑSyn.
From Cellular Triggers to Strain Maturation
Although ɑSyn has been historically viewed as a protein important for synaptic or neuronal function, it has multiple functional roles, since, as we described earlier, the protein is expressed in multiple cell types in tissue outside the central nervous system. ɑSyn is abundant in peripheral tissue of barrier organs and an increasing number of studies have shown that exposure to pathogens within these visceral sites can lead to accumulation of ɑSyn via infiltrating immune cells or alternatively, via triggering its expression in peripheral neurons as part of an innate immune response [21, 22, 38, 39, 42,43,44,45,46, 120]. Next to this cellular diversity, native ⍺Syn interacts with multiple subcellular structures and across cellular compartments and depending on its precise role it can be located within the synapse, the soma, or the nucleus of the cell [121].
To fulfill these distinct roles, the conformational flexibility of ɑSyn provides the protein with an advantage. However, because of its pleomorphic nature, the protein may also form unwanted interactions when its expression levels are triggered to levels that exceed its own solubility [59]. Inflammation and infections may trigger expression of ɑSyn, especially at visceral barrier sites, where pathogens can more freely interact with ɑSyn-expressing cells [42, 122]. Pesticides or infectious pathogens can directly stimulate the expression of ɑSyn in enteric or peripheral innervations [123, 124]. Viral infections have been associated with aggregated ɑSyn within peripheral sites [120, 125], but also in the brain during encephalitis with neurotropic agents [126, 127].
Multiple triggers could thus potentially lead to the aggregation of ɑSyn with a sustained proteopathic burden. Inflammation or infection triggered aggregation appears to be a general effect that could take place in any individual exposed to an environmental trigger. Aggregated ɑSyn has indeed been found in visceral sites, for instance in the gut or the appendix [128], of apparently normal individuals, without any signs of neurological illness. A popular hypothesis is that aggregated ɑSyn can escape these visceral sites and spread to the central nervous system via parasympathetic or sympathetic connections [22, 129]. Indeed, injection of fibrils in the gut of wild type mice and transgenic rats was shown to induce transmission to the brainstem via the vagal nerve after which it further propagates to distant areas [130, 131]. Parkinsonian risk factors or aging could further influence these effects, leading to a more permissive transfer of pathogenic ɑSyn from the periphery to the brain [132, 133]. Similarly, injection of fibrillar ɑSyn in the urinary tract was recently demonstrated to lead to its transmission to the brainstem via sympathetic and spinal projections [134].
Based on these observations, a hypothetical model can be constructed that leads to fibrillar maturation of phenotype-specific ɑSyn strains during in vivo transmission (Fig. 3). In this model, fibrillar assemblies will passage between multiple cells via their connectome and will be repeatedly subjected to distinct intracellular interactomes. The transmission route will depend on cell-autonomous factors such as the expression levels of ɑSyn, cellular or disease-specific risk factors, and the selective vulnerability of the recipient cell. For each cellular passage, the transmitted seed will be subjected to new cellular environmental conditions and amplify with heterogeneous ɑSyn folding intermediates. When ɑSyn filaments amplify with endogenous ɑSyn, cell-specific folding intermediates can incorporate their structural changes, inherent to the environment as their templating and the intracellular amplification of ɑSyn will directly depend on their cellular cofactors. Similarly, the quaternary arrangement of two filaments can also be affected since some cells might express certain cofactors required for the interaction of these filaments. By passaging and amplification within different environments, the conformation of the transmitted seed can be significantly impacted. Repeated passages between different cells, such as neurons or oligodendrocytes, could thus lead to the formation of distinct and mature ɑSyn strains that will not undergo anymore structural changes and have cellular or disease-specific post-translational modifications as opposed to the greater heterogeneity of transient assemblies en route to a more stable conformational state that can be maintained by the host. The conformational state of these mature assemblies will thereby reflect syndrome-specific transmission pathways between a trigger site and central structures with mature pathology.

A model of ɑSyn strain maturation during sequential in vivo transmission. A A trigger causes transient upregulation of ɑSyn, leading to its intracellular aggregation. Stable ɑSyn aggregates can transmit to neighboring cells via connected transcellular pathways. In the presence of sufficient endogenous ɑSyn in the recipient cell, amplification can occur with the transmitted seed. The transfer of ɑSyn fibrillar seeds to resilient cells or cells that lack sufficient endogenous ɑSyn will lead to unsuccessful amplification and halt serial transmission. Different cell types (A or B) can uniquely influence templated amplification as ɑSyn aggregates will interact with unique cellular risk factors. During in vivo amplification, cell-specific cofactors will differentially impact the conformational state of ɑSyn folding intermediates or ɑSyn filaments and introduce structural variations during templated amplification. The final conformation of the mature fibril is the function of the in vivo connectome, cellular ɑSyn expression, and cellular risk factors. B The conformational landscape of ɑSyn aggregates after sequential transmission via alternative transmission pathways. The maturation of ɑSyn fibrils from heterogeneous assemblies into phenotype-specific strains is shown over serial passages. An ɑSyn trigger elicits expression of ɑSyn, of which the assemblies lack a dominant conformation and this is reflected by the heterogeneous folding landscape of the populating assemblies. After transmission of ɑSyn seeds to the recipient cell, mature seeds will amplify intracellularly, whereas non-amplifying seeds will be degraded, leading to a narrowing of the conformational spectrum. Only a limited number of cell types will sustain intracellular amplification (positive selection) whereas other cell types will inhibit amplification (negative selection) resulting in a further structural selection. Templated amplification during sequential transmission will lead to the in vivo formation of mature ɑSyn fibrils and formation of a predominant strain in vivo
Conclusion
The conformational features of ɑSyn during health and disease have been a long-studied topic in synucleinopathies. The native assembly of ɑSyn into functional or folding intermediates in a rapidly changing environment needs to be strictly controlled to avoid the protein to form unwanted interactions. Inflammatory interactions at visceral barrier sites may trigger ɑSyn expression during high metabolic burden and lead to its assembly into potential pathogenic aggregates. In vivo transmission models have demonstrated how these aggregates can escape peripheral sites and transmit transcellularly to the central nervous system [12, 14]. It appears that ɑSyn transmission routes follow a spreading pattern determined by their cellular connectome and this spread is facilitated by cellular risk factors inherent to Lewy disorders or MSA.
By experimentally varying in vitro assembly conditions, ɑSyn aggregation into recombinant fibrils can yield a great variety of structural states. In contrast, the number of possible structural conformations of ɑSyn aggregates in human brain and in vivo seeding models appears to be much more restricted [5,6,7,8, 17, 104, 119]. There is also a significant structural overlap between ɑSyn filaments isolated from patient brain with a similar clinical diagnosis [3,4,5]. This illustrates that there is a remarkable and maybe unexpected biological uniformity between the conformations of fibrillar ɑSyn.
Even if it is not yet clear why such a disease-specific structural fingerprint exists, the cellular conditions in which ⍺Syn forms stable fibrils might be instrumental for these assemblies to form. This phenotype-specific conformational uniformity could point to a molecular signature of disease and as such, ɑSyn strains could potentially serve as a biological substrate of synucleinopathy subtypes. This has important but promising implications for ɑSyn biomarker-based diagnostics that are currently under development.
It remains to be investigated how structural information is translated onto elongating fibrils during seeded amplification in vivo. The cellular environment appears to have a significant impact on the in vivo assembly of fibrillar ɑSyn during sequential transmission. By comparing the structure of amplified and non-amplified fibrils from patient brain, it was shown that aggregation conditions can impact seeded amplification and that information might be partially lost during in vitro templating [6]. Although some phenotypic effects are translated and propagated robustly during fibrillar transmission in vivo, this discrepancy also reflects an important shortcoming of current preclinical models.
To improve the translational value of these models as well as to improve cross-laboratory findings, the effects of isolating and amplifying recombinant strains from native disease-specific assemblies for transmission studies need to be better understood. Within the same lines, it will be important to provide the correct ɑSyn substrate in vitro and in vivo, to allow faithful amplification of disease-specific conformations. This might require working with assemblies with disease-specific post-translational modifications or providing protein or membrane substrates for reliable templating during seeded amplification. Providing a physiological environment that is both permissive but also resilient for serial transmission and allowing sufficient time for assemblies to seed and transmit so that Lewy- or MSA-like pathology can develop in a progressive manner could promote the formation of mature, compact inclusions that more closely mimic those of the human brain. By standardizing analyses methods that leave aggregates and brain tissue intact, and that are fast to implement in the lab, for instance via in situ labeling, additional quality controls could ensure further standardization and improve the translational value of these preclinical transmission models. Hence, it will be important to determine what the exact contribution of the cellular environment is, and whether in vitro conditions can be further optimized so that experimental Lewy and MSA phenotypes can be more robustly reproduced.
Future studies will need to confirm that this structural uniformity of ɑSyn between diseases indeed exists, and if so, the field will need to devise new methods and guidelines for the standardization and development of recombinant disease-specific fibrils and their use for in vitro, in cellulo, and in vivo studies as slight variations between assembly conditions could significantly impact the experimental outcome. Doing so will further improve the validity of preclinical studies, potentially via humanizing animal models, removing the species barrier, and assuring full sequence homology for reliable templated seeding with human ɑSyn strains in vivo. Collectively, these new insights will facilitate the development of novel biomarkers and facilitate ɑSyn-targeting drug discovery efforts.
References
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet (London, England). 2011;377:641–9.
Goedert M, Jakes R, Spillantini MG. The synucleinopathies: twenty years on. J Parkinsons Dis. 2017;7:S51.
Grazia Spillantini M, Anthony Crowther R, Jakes R, et al. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998.
Schweighauser M, Shi Y, Tarutani A, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;1–21.
Lövestam S, Schweighauser M, Matsubara T, et al. Seeded assembly in vitro does not replicate the structures of a-synuclein filaments from multiple system atrophy.
Yang Y, Shi Y, Schweighauser M, et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature. 2022;610.
Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;1–23.
Bellomo G, De Luca CMG, Paoletti FP, et al. α-Synuclein seed amplification assays for diagnosing synucleinopathies. Neurology. 2022;99:195–205.
Coysh T, Mead S. The future of seed amplification assays and clinical trials. Front Aging Neurosci. 2022;14:488.
Marmion D, Peelaerts W, Kordower J. A historical review of multiple system atrophy with a critical appraisal of cellular and animal models. J Neural Transm. 2021;128:1507–27.
Henderson MX, Trojanowski JQ, Lee VMY. α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci Lett. 2019;709:134316.
Lashuel HA, Overk CR, Oueslati A, et al. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48.
Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener. 2020;15:1–12.
Peelaerts W, Bousset L, Baekelandt V, et al. ɑ-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences. Cell Tissue Res. 2018;373:195–212.
Peng C, Gathagan RJ, Covell DJ, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018.
Torre-Muruzabal T, Van der Perren A, Coens A, et al. Host oligodendrogliopathy and ɑ-synuclein strains dictate disease severity in multiple system atrophy. Brain. 2022.
Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;1–25.
Pande R, Huang Y, Teeple E, et al. Single cell atlas of human putamen reveals disease specific changes in synucleinopathies: Parkinson’s disease and multiple system atrophy.
Wilhelm BG, Mandad S, Truckenbrodt S, et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–8.
Schaeffer E, Kluge A, Böttner M, et al. Alpha synuclein connects the gut-brain axis in Parkinson’s disease patients – a view on clinical aspects, cellular pathology and analytical methodology. Front Cell Dev Biol. 2020;8:910.
Borghammer P, Van Den Berge N. Brain-first versus gut-first Parkinson’s disease: a hypothesis. J Parkinsons Dis. 2019;9:S281–95.
Barbour R, Kling K, Anderson JP, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis. 2008;5:55–9.
Bartels T, Ahlstrom LS, Leftin A, et al. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys J. 2010;99:2116.
Man WK, Tahirbegi B, Vrettas MD, et al. The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition. Nat Commun. 2021;12:1–10.
Lautenschläger J, Kaminski CF, Schierle GSK. α-Synuclein, regulator of exocytosis, endocytosis, or both? Trends Cell Biol. 2017;27:468–79.
Lautenschläger J, Stephens AD, Fusco G, et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat Commun. 2018;9(1):1–13.
Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79:1044–66.
Devi L, Raghavendran V, Prabhu BM, et al. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–100.
Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim Biophys Acta - Biomembr. 2012;1818:162–71.
Hallacli E, Kayatekin C, Nazeen S, et al. The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell. 2022;185:2035-2056.e33.
Burré J, Sharma M, Südhof TC. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med. 2018;8.
Burré J, Sharma M, Südhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–83.
Liu C, Zhao Y, Xi H, et al. The membrane interaction of alpha-synuclein. Front Cell Neurosci. 2021;15:30.
Dikiy I, Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta. 2012;1818:1013.
Kim KS, Park JY, Jou I, et al. Regulation of Weibel-Palade body exocytosis by α-synuclein in endothelial cells. J Biol Chem. 2010;285:21416.
Tamo W, Imaizumi T, Tanji K, et al. Expression of α-synuclein, the precursor of non-amyloid β component of Alzheimer’s disease amyloid, in human cerebral blood vessels. Neurosci Lett. 2002;326:5–8.
Grozdanov V, Danzer KM. Intracellular alpha-synuclein and immune cell function. Front Cell Dev Biol. 2020.
Kasen A, Houck C, Burmeister AR, et al. Upregulation of α-synuclein following immune activation: possible trigger of Parkinson’s disease. Neurobiol Dis. 2022;166.
Fagerberg L, Hallstrom BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406.
Pei Y, Maitta RW. Alpha synuclein in hematopoiesis and immunity. Heliyon. 2019;5.
Tulisiak CT, Mercado G, Peelaerts W, et al. Can infections trigger alpha-synucleinopathies? Mol Biol Neurodegener Dis Visions Futur Part A. Elsevier; 2019. p. 299–322.
Alam MM, Yang D, Li XQ, et al. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022;38.
Monogue B, Chen Y, Sparks H, et al. Alpha-synuclein supports type 1 interferon signalling in neurons and brain tissue. Brain. 2022.
Beatman EL, Massey A, Shives KD, et al. Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol. 2015;90:2767–82.
Massey AR, Beckham JD. Alpha-synuclein, a novel viral restriction factor hiding in plain sight. DNA Cell Biol. 2016.
Moreno-Valladares M, Moncho-Amor V, Bernal-Simon I, et al. Norovirus intestinal infection and Lewy body disease in an older patient with acute cognitive impairment. Int J Mol Sci. 2022;23.
Uversky VN, Eliezer D. Biophysics of Parkinson’s disease: structure and aggregation of α-synuclein. Curr Protein Pept Sci. 2009;10:483.
Fink AL. The aggregation and fibrillation of α-synuclein. Acc Chem Res. 2006;39:628–34.
Alam P, Bousset L, Melki R, et al. α-Synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem. 2019;150:522–34.
Ghosh D, Mehra S, Sahay S, et al. α-Synuclein aggregation and its modulation. Int J Biol Macromol. 2017;100:37–54.
Brown JWP, Meisl G, Knowles TPJ, et al. Kinetic barriers to α-synuclein protofilament formation and conversion into mature fibrils. Chem Commun. 2018;54:7854–7.
Cremades N, Cohen SIA, Deas E, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149:1048–59.
Gaspar R, Meisl G, Buell AK, et al. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. 2017;50:e6.
Cremades N, Chen SW, Dobson CM. Structural characteristics of α-synuclein oligomers. Int Rev Cell Mol Biol. 2017;329:79–143.
Chen SW, Drakulic S, Deas E, et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci U S A. 2015;112:E1994–2003.
Cascella R, Chen SW, Bigi A, et al. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat Commun. 2021;12:1–16.
Fusco G, Chen SW, Williamson PTF, et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science. 2017;358:1440–3.
Breydo L, Wu JW, Uversky VN. α-Synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta - Mol Basis Dis. 2012;1822:261–85.
Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575.
Mahul-Mellier AL, Burtscher J, Maharjan N, et al. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 2020;117:4971–82.
Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012;209:975–86.
Luk KC, Kehm V, Carroll J, et al. Pathological α-synuclein transmission in nontransgenic mice. Science. 2012;338:949–53.
Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136:1128.
Rey NL, Steiner JA, Maroof N, et al. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016;213:1759–78.
Koller EJ, Brooks MMT, Golde TE, et al. Inflammatory pre-conditioning restricts the seeded induction of α-synuclein pathology in wild type mice. Mol Neurodegener. 2017;12:1–13.
Paumier KL, Luk KC, Manfredsson FP, et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–99.
Osterberg VR, Spinelli KJ, Weston LJ, et al. Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–60.
Froula JM, Castellana-Cruz M, Anabtawi NM, et al. Defining α-synuclein species responsible for Parkinson’s disease phenotypes in mice. J Biol Chem. 2019;294:10392–406.
Peelaerts W, Bousset L, Van Der Perren A, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015.
Abdelmotilib H, Maltbie T, Delic V, et al. ⍺-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiol Dis. 2017;105:84–98.
Rey NL, George S, Steiner JA, et al. Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 2018;135:65–83.
Luna E, Decker SC, Riddle DM, et al. Differential α-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018;135:855.
Guo JL, Covell DJ, Daniels JP, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103.
Kim C, Lv G, Lee JS, et al. Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci Rep. 2016;6.
Ferreira N, Gram H, Sorrentino ZA, et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021;142:87–115.
Lau A, So RWL, Lau HHC, et al. α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci. 2020;23:21.
Liu D, Guo JJ, Su JH, et al. Differential seeding and propagating efficiency of α-synuclein strains generated in different conditions. Transl Neurodegener. 2021;10:1–15.
Woerman AL, Oehler A, Kazmi SA, et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019;137:437–54.
Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308–17.
Van Der Perren A, Gelders G, Fenyi A, et al. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020;139:977–1000.
Watts JC, Giles K, Oehler A, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013;110:19555–60.
Recasens A, Dehay B, Bové J, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62.
Bernis ME, Babila JT, Breid S, et al. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun. 2015;1–18.
Dhillon JKS, Trejo-Lopez JA, Riffe C, et al. Comparative analyses of the in vivo induction and transmission of α-synuclein pathology in transgenic mice by MSA brain lysate and recombinant α-synuclein fibrils. Acta Neuropathol Commun. 2019;7:80.
Mougenot AL, Nicot S, Bencsik A, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–8.
Frieg B, Geraets JA, Strohäker T, et al. Quaternary structure of patient-homogenate amplified α-synuclein fibrils modulates seeding of endogenous α-synuclein. Commun Biol. 2022;5:1–10.
Strohäker T, Jung BC, Liou SH, et al. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat Commun. 2019;10:1–12.
Tanudjojo B, Shaikh SS, Fenyi A, et al. Phenotypic manifestation of α-synuclein strains derived from Parkinson’s disease and multiple system atrophy in human dopaminergic neurons. Nat Commun. 2021;12:1–16.
Killinger BA, Marshall LL, Chatterjee D, et al. In situ proximity labeling identifies Lewy pathology molecular interactions in the human brain. Proc Natl Acad Sci U S A. 2022;119:e2114405119.
Shahmoradian SH, Lewis AJ, Genoud C, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22:1099–109.
Moors TE, Maat CA, Niedieker D, et al. The subcellular arrangement of alpha-synuclein proteoforms in the Parkinson’s disease brain as revealed by multicolor STED microscopy. Acta Neuropathol. 2021;142:423–48.
Zhang S, Zhu R, Pan B, et al. Post-translational modifications of soluble α-synuclein regulate the amplification of pathological α-synuclein. Nat Neurosci. 2023;26:213–25.
Ghanem SS, Majbour NK, Vaikath NN, et al. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc Natl Acad Sci U S A. 2022;119:e2109617119.
Ma MR, Hu ZW, Zhao YF, et al. Phosphorylation induces distinct alpha-synuclein strain formation. Sci Rep. 2016;6:1–11.
Henderson MX, Cornblath EJ, Darwich A, et al. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci. 2019;22:1248–57.
Henrich MT, Geibl FF, Lakshminarasimhan H, et al. Determinants of seeding and spreading of α-synuclein pathology in the brain. Sci Adv. 2020;6.
Mezias C, Rey N, Brundin P, et al. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: involvement of retrograde and anterograde axonal propagation. Neurobiol Dis. 2020;134:104623–9.
Volpicelli-Daley LA, Abdelmotilib H, Liu Z, et al. G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J Neurosci. 2016;36:7415.
Bieri G, Brahic M, Bousset L, et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019;137:961.
Henderson MX, Sedor S, McGeary I, et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron. 2020;105:822-836.e7.
Migdalska-Richards A, Wegrzynowicz M, Harrison IF, et al. L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse α-synuclein pre-formed fibrils. PLoS ONE. 2020;15:e0238075.
Johnson ME, Bergkvist L, Stetzik L, et al. Heterozygous GBA D409V and ATP13a2 mutations do not exacerbate pathological α-synuclein spread in the prodromal preformed fibrils model in young mice. Neurobiol Dis. 2021;159:105513.
Klingstedt T, Ghetti B, Holton JL, et al. Luminescent conjugated oligothiophenes distinguish between α-synuclein assemblies of Parkinson’s disease and multiple system atrophy. Acta Neuropathol Commun. 2019;7:1–9.
Choi ML, Chappard A, Singh BP, et al. Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat Neurosci. 2022;25:1134–48.
Wakabayashi K, Tanji K, Odagiri S, et al. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2012;47:495–508.
Erskine D, Koss D, Korolchuk VI, et al. Lipids, lysosomes and mitochondria: insights into Lewy body formation from rare monogenic disorders. Acta Neuropathol. 2021;141:511.
Klingstedt T, Åslund A, Simon RA, et al. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org Biomol Chem. 2011;9:8325–56.
Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Wenning GK, Seppi K, Jellinger K. A novel grading scale for striatonigral degeneration (multiple system atrophy). J Neural Transm. 2002;1–14.
Laferrière F, Claverol S, Bezard E, et al. Similar neuronal imprint and no cross-seeded fibrils in α-synuclein aggregates from MSA and Parkinson’s disease. NPJ Park Dis. 2022;8:1–12.
Ishii A, Dutta R, Wark GM, et al. Human myelin proteome and comparative analysis with mouse myelin. Proc Natl Acad Sci U S A. 2009;106:14605–10.
Fitzner D, Bader JM, Penkert H, et al. Cell-type- and brain-region-resolved mouse brain lipidome. Cell Rep. 2020;32.
Asi YT, Simpson JE, Heath PR, et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014.
Miller DW, Johnson JM, Solano SM, et al. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm. 2005;112:1613–24.
Dulken BW, Buckley MT, Negredo PN, et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches.
Kenigsbuch M, Bost P, Halevi S, et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat Neurosci. 2022;25:876–86.
Jäkel S, Agirre E, Mendanha Falcão A, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019.
Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020.
Labrie V, Brundin P. Alpha-synuclein to the rescue: immune cell recruitment by alpha-synuclein during gastrointestinal infection. J Innate Immun. 2017;9:437–40.
Miraglia F, Ricci A, Rota L, et al. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen Res. 2018;13:1136.
Linard M, Ravier A, Mougué L, et al. Infectious agents as potential drivers of α-synucleinopathies. Mov Disord. 2022;37:464–77.
Pan-Montojo F, Schwarz M, Winkler C, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Reports. 2012;2:1–12.
Sampson TR, Challis C, Jain N, et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife. 2020;9:319–96.
Follmer C. Viral infection-induced gut dysbiosis, neuroinflammation, and α-synuclein aggregation: updates and perspectives on COVID-19 and neurodegenerative disorders. ACS Chem Neurosci. 2020;11:4012–6.
Ait Wahmane S, Achbani A, Ouhaz Z, et al. The possible protective role of α-synuclein against the SARS-CoV-2 infections in patients with Parkinson’s disease. Mov Disord. 2020;mds.28185-5.
Olsen LK, Dowd E, McKernan DP. A role for viral infections in Parkinson’s etiology? Neuronal Signal. 2018;2:314–7.
Killinger BA, Madaj Z, Sikora JW, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. 2018;10.
Johnson ME, Stecher B, Labrie V, et al. Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci. 2019;42:4–13.
Kim S, Kwon S-H, Kam T-I, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627-641.e7.
Van Den Berge N, Ferreira N, Gram H, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138:535–50.
Van Den Berge N, Ferreira N, Mikkelsen TW, et al. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain. 2021;144:1853.
Challis C, Hori A, Sampson TR, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. 2020;1–27.
Ding X, Zhou L, Jiang X, et al. Propagation of pathological α-synuclein from the urogenital tract to the brain initiates MSA-like syndrome. iScience. 2020;23:101166.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
We acknowledge financial support from the FWO-Vlaanderen (postdoctoral fellowship to WP and project G081121N) and the KU Leuven (project C14/18/102).
Author information
Authors and Affiliations
Contributions
WP conceptualized and wrote the manuscript; VB corrected and finalized the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Peelaerts, W., Baekelandt, V. ⍺-Synuclein Structural Diversity and the Cellular Environment in ⍺-Synuclein Transmission Models and Humans. Neurotherapeutics 20, 67–82 (2023). https://doi.org/10.1007/s13311-023-01365-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-023-01365-5