
Abstract
Laryngeal squamous cell carcinoma (LSCC) is a very aggressive cancer, considered to be a subtype of the head and neck squamous cell carcinoma (HNSCC). Despite significant advances in the understanding and treatment of cancer, prognosis of patients with LSCC has not improved recently. In the present study, we sought to understand better the genetic mechanisms underlying LSCC development. Thirty-two tumor samples were collected from patients undergoing surgical resection of LSCC. The samples were submitted to whole-genome cDNA microarray analysis aiming to identify genetic targets in LSCC. We also employed bioinformatic approaches to expand our findings using the TCGA database and further performed functional assays, using human HNSCC cell lines, to evaluate viability, cell proliferation, and cell migration after silencing of selected genes. Eight members of the homeobox gene family (HOX) were identified to be overexpressed in LSCC samples when compared to normal larynx tissue. Quantitative RT-PCR analysis validated the overexpression of HOX gene family members in LSCC. Receiver operating characteristic (ROC) statistical method curve showed that the expression level of seven members of HOX gene family can distinguish tumor from nontumor tissue. Correlation analysis of clinical and gene expression data revealed that HOXC8 and HOXD11 genes were associated with the differentiation degree of tumors and regional lymph node metastases, respectively. Additionally, siRNA assays confirmed that HOXC8, HOXD10, and HOXD11 genes might be critical for cell colony proliferation and cell migration. According to our findings, several members of the HOX genes were overexpressed in LSCC samples and seem to be required in biological processes involved in tumor development. This suggests that HOX genes might play a critical role in the physiopathology of LSCC tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Laryngeal squamous cell carcinoma (LSCC) is one of the 15 most common types of cancer in men worldwide [1]. Despite recent advances in cancer treatment, this type of malignancy has one of the lowest survival rates among major cancer types [2]. Tobacco and alcohol consumption are major risk factors involved in LSCC development [2]. Histopathological tumor classification and TNM staging system are currently the predominant methods used to predict disease outcome in LSCC patients [3].
Although previous studies have addressed the molecular characterization of LSCC [4–6], our understanding of target genes involved in the biology of LSCC is still limited. Therefore, there is a need for new efforts to improve the characterization of molecular mechanisms involved in LSCC. This is especially important since new therapies interfering in specific targets within genetic pathways may become available, as demonstrated for other cancer types [7–10]. In the present study, we performed a molecular analysis in a series of 32 human LSCC samples aiming to identify genes involved in the pathogenesis of LSCC. We employed gene expression profiling approach along with ample statistical analysis [11] to show that LSCC is marked by a series of transcriptional alterations, including overexpression of eight HOX gene family members. Increased expression of HOX gene members in LSCC was validated by RT-qPCR. Knockdown of HOXC8, HOXD10, or HOXD11 expression indicates their involvement in biological processes related to LSCC development. In summary, the results suggest that HOX family members play a significant role in the physiopathology of LSCC tumors.
Material and methods
Ethics statement and tissue sample collections
This study was approved by the Ethics Committee of Ribeirão Preto Medical School, University of São Paulo (USP) (Proc. No. 9371/2003). Written informed consent was obtained from patients undergoing surgical resection at the Head and Neck Surgery Division of the Department of Ophthalmology, Otorhinolaryngology and Head & Neck of Ribeirão Preto Medical School, USP (Brazil), from January 2005 to December 2009. Inclusion criteria were histopathological diagnosis of LSCC and elective surgeries for LSCC in patients without previous treatments and patients’ allowance to donate part of their tumor for genetic studies. Exclusion criteria were doubtful diagnosis of LSCC, unavailable postsurgical follow-up, and patients without complete clinical data or signed agreement for collection of samples. A total of 32 patients could be included in this study. After histopathological confirmation of LSCC and microdissection of tumors from their surgical safety border (normal tissue; n = 13), tissue samples were snap-frozen in liquid nitrogen.
Microarray experiments
First, RNA was purified with RNeasy kit (Qiagen, Valencia, CA, USA) and quantified with the NanoDrop spectrophotometer (260 nm; Thermo Fisher Scientific, Waltham, MA, USA). Agarose gel electrophoresis was used to evaluate the quality of the RNAs (28S and 18S ribosomal RNA detection). We used Fluidics Station 450 system (Affymetrix) and the Quick Amp Labeling One-Color kit (5190–0442; Agilent, USA) for RNA hybridization to Whole Human Genome Oligo Microarray chips (G4112F, Agilent Technologies, Santa Clara, CA, USA). One Color RNA Spike-In Kit (5188–5282; Agilent, USA) was used for loading control. Each array reflected the expression of a single sample, and the CEL files from scanned microarrays were produced using the GenePix 4000B scanner (Axon Instruments, USA) together with the GenePix Pro 6.0 and Agilent Feature Extraction 9.5.3.1 software. Raw microarray data were deposited at Gene Expression Omnibus (GEO) (accession ID: GSE59102) and published in Genomic Data [11].
Microarray data analysis
Data analysis was performed with R packages from the Bioconductor Project (www.bioconductor.org). Spearman rank coefficient-based correlation and average linkage methods were applied for hierarchical clustering and exclusion of masked spots from microarray dataset. Statistical significance was determined with a nonpaired t test analysis. False discovery rate (FDR) was used to adjust p values (q values). A heat map was generated to illustrate the results.
TCGA data analysis
Differential expression analysis between head and neck squamous cell carcinoma (HNSCC), and normal samples from the same patient was performed using the bioconductor R package TCGA Biolinks [12]. p Values were adjusted for FDR <0.05 as multiple hypothesis test correction method. Genes with log2 fold change less than −2 or greater than 2 and adjusted p value <0.01 were considered differentially expressed.
RT-qPCR analysis
High Capacity cDNA Reverse Transcription kit (Applied Biosystems, USA) was used to generate cDNAs from 1 μg of extracted RNA according to the manufacturers’ instructions. Then, cDNAs were diluted at 1:5 and stored at −80 °C until analysis. RT-qPCR was performed with PrimeTime® Mini qPCR Probe kit (Integrated DNA Technologies, USA). For all RT-qPCR reactions, median geometric expression from housekeeping GAPDH and TBP genes were used to normalize RNA inputs. The levels of expressed genes were measured by RT-qPCR using the 2−ΔΔCt method [13].
Cell culture
FADU and UMSCC14 cell lines from human head and neck squamous cell carcinomas were used to functional assays. Both were kindly provided by Dr. Eloiza H. Tajara of the School of Pharmaceutical Sciences–UNESP. Cell lines were cultured under standard conditions in Minimum Essential Medium (Gibco®) and Dulbecco’s modified Eagle’s medium (DMEM, Gibco®), respectively, supplemented with 10 % fetal bovine serum and 1 % penicillin and streptomycin. Cells were kept at 37 °C and 5 % CO2.
siRNA assay
HOXC8 (siHOXC8), HOXD10 (siHOXD10), and HOXD11 (siHOXD11) siRNAs and negative control siRNA (siCTRL) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cells were transfected with specific siRNAs (30 nM final concentration) using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. Subsequent experiments were performed 48 h after transfection.
Cell viability
Cell viability and apoptosis were evaluated using Annexin V-FITC apoptosis detection kit (Becton Dickinson, Holdrege, NE, USA). All experiments were performed according to the manufacturer’s guidelines. FACSCalibur flow cytometer was used for all analysis (Becton Dickinson, Franklin Lakes, NJ, USA). Double-negative cells were considered viable. Annexin V-positive cells were determined as apoptotic cells. Results were shown as percentage of positive stained cells.
Colony formation assay
Cell suspension was settled into six-well plates (500 cells/well). After 12 days, cells were fixed in 4 % formaldehyde (in PBS) and stained with 0.5 % crystal violet. Number of colonies was counted with ImageJ software.
Transwell migration assay
Cell motility was evaluated in 24-well transwell plates (Greiner, USA). FADU and UMSCC14 cells (1 × 105cells/300 μL serum-free medium) were seeded in the upper chambers of the transwell plates 48 h after transfection with individual siRNAs. The lower chambers were filled with 500 μL of medium added 10 % FBS. Cells were allowed to migrate for 24 h at 37 °C. Then, the cells from the upper compartment were removed with a cotton swab, and the cells that migrate to the lower face of the filter were fixed in 4 % formaldehyde (in PBS) and stained with 0.5 % crystal violet. The number of cells was manually counted using ImageJ software.
Statistical analysis
Data were analyzed using GraphPad Prism 5.0 software package (Graph Pad Software Inc., USA). Statistical significance was determined by one-way ANOVA followed by Bonferroni post hoc test. Mann-Whitney’s test was applied for comparisons between two-independent groups. IBM SPSS Statistics for Mac (version 20.0) was used for analysis of survival (Kaplan-Meier’s test), receiver operating characteristic (ROC) curve analysis and categorical data classification (Fisher’s test). A probability of p < 0.05 was considered to be statistically significant. All data are shown as mean ± standard deviation.
Results
Clinical characteristics of the studied population
Patients were predominately male (31/32) with a history of smoking (32/32) and alcohol abuse (31/32) (Table 1 and Suppl. Table 1). From 32 patients, 8 (25 %) suffered tumor relapse and 3/32 (9.4 %) tumor metastasis. A single patient suffered tumor relapse followed by distant organ metastasis. Two patients died 1 week after tumor surgery (2/12, 16.7 %), two other patients (2/12, 16.7 %) died from noncancer related reasons, and 8/12 (66.6 %) died from cancer. Among the 20 survivors, four patients had tumor relapse, and one patient suffered from metastatic disease. Thus, tumor relapse and metastasis were determining features impacting patient survival (Suppl. Fig. 1).
Microarray analysis in LSCC
To investigate potential genetic targets and biomarkers in LSCC, microarray analysis was performed from surgical samples comprising of tumor as well as nonneoplastic tissue. Statistical analysis restricted to those transcripts with log2 fold change higher than 3.2 and p ≤ 1e−7 revealed clear transcriptomic differences between nontumor and tumor samples, with 70 genes differentially expressed. The volcano plot in Fig. 1a highlights the group of 70 genes differentially expressed between normal and tumor tissues, from which 30 genes (42 %) were significantly overexpressed in LSCC (Suppl. Table 2). Figure 1b shows the heat map of the hierarchical clustering using the expression pattern of this group of 70 genes differentially expressed. Interestingly, we noticed that eight members of the HOX gene family were overexpressed in LSCC. This transcription factor family was the only genetic family found overexpressed as a cluster in LSCC samples (Fig. 1b), and this enrichment was confirmed by a gene enrichment analysis (Ingenuity Systems, USA). On average, HOX gene family members showed an overexpression of 4.2 (±0.7)-fold in tumors in comparison with normal tissue (surrounding tumor).

Microarray analysis in laryngeal squamous cell carcinoma (LSCC). (a) Volcano plot depicts 70 genes (green and red dots) differentially expressed between LSCC and nontumoral tissues. X-axis represents fold change values (FC; log2) and Y-axis p value (− log10). A threshold cutoff (3.32-FC vs p value 10−7) was applied for selection of genes downregulated (green dots/right side) or upregulated (red dots/left side) in LSCC samples. (b) Two-way unsupervised hierarchical clustering is illustrated in a heat map diagram. Each column represents a sample, and each row indicates a gene. Sample clustering tree is shown at the top, while that for genes appears on the left. Across all samples, relative gene expression levels are shown in color scales (red, above the mean; green, lower than the mean; black, median expression). Eight members of HOX family are highlighted in red on the right side

Validation of transcriptomic data by RT-qPCR. Analysis was performed for HOXC8 (a), HOXC9 (b), HOXC10 (c), HOXC13 (d), HOXD10 (e), HOXD11 (f), HOXA10 (g), and HOXA11-AS1 genes (h). Relative gene expression calculations were performed according to the 2–∆∆Ct method using the mean Ct values of GAPDH and TBP genes as endogenous control and the mean value of normalized Cts of all normal samples as reference. Mann-Whitney’s test was used for statistical analysis (*p < 0.05)
Validation of overexpression of HOX gene members in LSCC
Gene expression analysis by RT-qPCR confirmed that all eight members of HOX gene family were overexpressed in tumors in comparison with normal tissue (Fig. 2 and Suppl. Table 3). In general, the expression changes detected by RT-qPCR were even more dramatic than those observed in the microarray analysis. The relative expression level (fold change) of the four HOXC gene members between LCSS and normal tissue varied from 6.4- to 44.6-fold (Fig. 2a–d). For the members of HOXD group, overexpression means of 6.4-fold (HOXD10) and 38.7-fold (HOXD11) were detected in LSCC samples (Fig. 2e, f). HOXA10 and HOXA11-AS1 were found 3.4-fold and 7.9-fold overexpressed in LSCC, respectively (Fig. 2g, h).

Volcano plot showing the comparison of gene expression between normal margin and head and neck squamous cell carcinoma (HNSCC) samples from TCGA database. X-axis represents log2 fold change values, and Y-axis shows − log10-adjusted p value. Red dots: HOX genes upregulated in TCGA database HNSCC samples. Purple dots highlight HOX genes found upregulated in HNSCC samples and in our cohort of LSCC samples as well
HOX genes expression in HNSCC based on TCGA database
Using the public TCGA database, we investigated the expression levels of all members of the HOX genes family (39 genes) in HNSCC tissues, which include samples of squamous cell carcinoma of tongue (13 samples), larynx (12 samples), oral cavity (13 samples), base of the tongue (2 samples), and floor of the mouth (3 samples) and their respective adjacent normal tissues. Fourteen HOX genes were identified as overexpressed in HNSCC tissues, from which six matched those found overexpressed in LSCC in our study (Fig. 3). One could suggest that HOX genes may be overexpressed as a cluster in HNSCC tissues by amplification events. The occurrence of genomic amplification in the seven HOX gene loci was evaluated using TCGA copy number variation (CNV) data [14]. We observed that the expression of most of the HOX genes that we found upregulated in LSCC had no correlation with copy number alterations (Table 2). The exception was HOXA10 that showed a weak correlation (R = 0.2). Furthermore, we searched the TCGA database for methylation status of those seven genes [15], and we found that expression levels and methylation status were inversely correlated (Spearman correlation less than or equal to −0.5) for HOXA10, HOXC10, and HOXD11 (Table 3), indicating that DNA methylation could be involved in the regulation of these HOX genes in HNSCC.
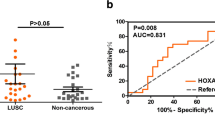
Expression level of HOX genes can contribute to distinguish tumor tissues from their normal counterparts
To evaluate if expression of HOX gene family members could discriminate between normal and tumor tissues, we performed a ROC curve analysis. Calculating the sensibility and specificity enabled us to determine an expression fold change cutoff for all HOX genes analyzed here, from which AUC and p value were determined. AUC > 0.8 values were found in six out of seven HOX genes indicating the value of those HOX genes as diagnostic markers (Table 4). We then investigated the association between the expression of the seven HOX genes and patient clinic pathologic data. HOXC8 gene overexpression was inversely associated with the degree of tumor differentiation (Table 5), while the presence of regional lymph node metastasis was significantly associated with HOXD11 gene overexpression (Table 6). The other members of HOX gene family did not show any correlation to clinical data, such as tumor size, differentiation degree, and regional or distant metastasis (Supplemental 4 to 8). For Kaplan-Meier analysis, the expression cutoff values from Table 4 were also used to separate samples into higher and lower expression groups for each of the HOX genes. However, no correlation between HOX gene expression levels and patients’ overall survival was observed for any of the seven analyzed genes (Suppl. Fig. 2).
HOXC8, HOXD10, and HOXD11 modulate cell survival, proliferation and migration
For functional assays, FADU and UMSCC14 cell lines were used, as they are HNSCC-derived cell lines expressing HOX genes. The efficiency of gene knockdown was evaluated 48 h after transfection with siRNAs for HOXC8, HOXD10, and HOXD11 by RT-qPCR (Suppl. 3A), and cell viability and apoptosis were quantified by flow cytometry (Suppl. Fig. 3B, C). HOXC8 knockdown altered cell viability and significantly increased apoptosis in FADU cells but had no effect on UMSCC14 cell viability or apoptosis rates (Suppl. Fig. 3B, C). On the other hand, neither HOXD10 nor HOXD11 knockdown caused any changes on survival rates of FADU or UMSCC14 cell lines (Suppl. Fig. 3). Colony formation assay demonstrated that knockdown of HOXC8, HOXD10, and HOXD11 genes in FADU and UMSCC14 cells significantly reduced their ability to establish colonies after 12 days of culturing (Fig. 4a, b).

Colony formation assay in FADU and UMSCC-14 cells after knockdown of HOXC8, HOXD10, or HOXD11 gene expression. a Representative images showing colonies formed by FADU cells (on the left) and UMSCC-14 cells (on the right) after 12 days of transfection with control siRNA (siCTRL) or HOXC8, HOXD10, or HOXD11 siRNAs. b Quantitation of colonies using ImageJ counting plugin. One-way ANOVA followed by Bonferroni’s post hoc test were used for statistical analysis (*p < 0.05). Each experiment was performed three times and each time in triplicate
Additionally, to further investigate the biological role of HOXC8, HOXD10, and HOXD11 in FADU and UMSCC14 cells, we performed a transwell migration assay 48 h after transfecting them with the respective siRNAs. Cells were allowed to migrate for 24 h, when we observed that reduced expression of any of the three HOX genes dramatically decreased cell migration in FADU cells and UMSCC-14 cells (Fig. 5a, b).

Cell migration assay in FADU and UMSCC-14 cells after knockdown of HOXC8, HOXD10, and HOXD11 genes. a Representative images from the bottom surface of the transwell insert, showing stained migrating cells: FADU (upper panel) and UMSCC-14 (bottom panel). b Bar graph displaying the percentage of migrating cells relative to the control (considered 100 %); FADU (left) and UMSCC-14 (right). One-way ANOVA followed by Bonferroni’s post hoc test was used for statistical analysis. Each experiment was performed three times and each time in triplicate
Discussion
In this study, we used mRNA microarray to investigate gene expression signatures associated with laryngeal squamous cell carcinoma progression. The analysis in 32 laryngeal tumor samples and 13 adjacent normal tissues identified 70 differentially expressed genes. A set of 30 genes was overexpressed in tumor samples, from which we highlighted a group of eight members of the HOX gene family (HOXA10, HOXA11-S1, HOXC8, HOXC9, HOXC10, HOXC13, HOXD10, and HOXD11). To our knowledge, this was the first observation of this relatively large number of HOX genes overexpressed simultaneously in a tumor type. RT-qPCR validation confirmed an impressive upregulation of all eight HOX genes in tumor samples compared to normal tissue, reaching around 40-fold in the case of the HOXC13 gene (Fig. 2). Numerous examples of individual aberrant HOX gene expression have been found in cancer. The mechanisms of this deregulation are multiple and can be tissue-dependent or temporal-spatially controlled [16].
Methylation of CpG islands in the promoter region of genes acts as a significant mechanism of epigenetic gene silencing in tumors, whereas demethylation can take place to upregulate oncogenes. Analysis of methylation status of seven HOX genes upregulated in LSCC using the TCGA database showed an inverse correlation between expression levels and methylation status of HOXA10, HOXC10, and HOXD11, indicating that this could be the mechanism responsible for the regulation of these three HOX genes in HNSCC. Aberrant DNA methylation patterns in a group of genes, including some HOX genes, were also found in human breast and ovarian cancer, and melanoma [17–19], suggesting that this could be the major mechanism of HOX gene regulation in tumors, possibly enabling tumorigenesis.
Expression of HOX genes is variable across different tumor types. Although some HOX genes were described as overexpressed in a particular tumor type, other reports have shown their downregulation in a different type of tumor [20]. This variation in the expression profile among different tumors indicates HOX genes can play either an oncogenic or tumor-suppressive function depending on the tumor type. HOXD10, for instance, was shown to be downregulated in gastric cancer and it was considered as a tumor suppressor candidate, since reexpression of HOXD10 in gastric cancer cell lines resulted in significant inhibition of cell survival, induction of apoptosis, and reduced cell migration and invasion [21]. Evidence of altered expression of HOXD10 has been also described in breast cancer, endometrial cancer, and hepatocellular carcinoma [22–25] in which HOXD10 expression is reduced in epithelial cells, as malignancy increases, or it is completely depleted in tumors compared to normal tissue. Whereas these findings suggest that HOXD10 has tumor-suppressive functions, a different scenario is observed for HNSCC. In the present study, although no correlation between expression of HOXD10 and the overall survival rate was detected, there was a significant upregulation in LSCC samples in comparison to normal tissues. This is in agreement with others studies that revealed significantly higher expression levels of HOXD10 in HNSCC and OSCC [26, 27]. Sharpe et al. [26], for instance, described HOXD10 as overexpressed in HNSCC tissues (oral cavity, oropharyngeal, pharyngeal SCC) and cell lines (tongue and pharynx SCC), and its knockdown resulted in impairment of cell proliferation and invasion. Our results also confirm the importance of HOXD10 for cell proliferation and migration.
HOXD11 was found downregulated through a specific methylation pattern of a group of genes in breast and ovarian cancer [17, 18] and melanoma [19]. Here, we found HOXD11 upregulated in LSCC samples compared to normal tissue, as it was found by Sharp et al. and Rodini et al. who described HOXD11 overexpressed in HNSCC and OSCC [26, 27], suggesting an important role for this gene in the development of these diseases. In addition, we showed data suggesting that HOXD11 contributes to tumor cell proliferation and migration.
HOXC8 also presented a variable pattern of expression. Immunohistochemical staining for HOXC8 was stronger in normal margin tissue than in pancreatic ductal adenocarcinoma tissue, and it was described as inversely related to progression and metastasis in this type of cancer [28]. Nevertheless, HOXC8 was reported as overexpressed in esophageal SCC and suggested as a potential prognostic marker for this type of cancer [29]. Similarly, we identified high expression levels of HOXC8 associated with lower tumor differentiation.
Other HOX genes identified herein were also described deregulated in different types of tumor. Although, the expression levels of most of them did not influence the overall survival rates, the members of the HOX family identified as overexpressed in LSCC in this study, except by HOXA10, contributed to distinguish tumor tissue from the normal surroundings.
HOX genes encode transcription factors and are known for acting in embryogenesis and regulating biological processes such as cell differentiation, cell proliferation, and apoptosis, which are important mechanisms for tissue and organ development [16]. According to our results, it is possible to infer that the genetic program regulated by these eight HOX genes would be reactivated in laryngeal SCC and could be associated with tumor development. Our findings suggested that HOXC8, HOXD10, and HOXD11 are associated with increased tumor cell proliferation and migration, whereas HOXC8 would in addition be involved in regulating cell survival. This is in agreement to other studies that reported a decrease in cell proliferation, migration, or invasion after depletion of HOXC8 [30], HOXD10, and HOXD11 [26] in breast cancer and HNSCC, respectively.
Conclusion
This study identified differentially expressed genes through a microarray genome-wide screening in LSCC samples. The results presented here revealed that eight members of the HOX gene family (HOXA10, HOXA11-S1, HOXC8, HOXC9, HOXC10, HOXC13, HOXD10, and HOXD11) were significantly upregulated in LSCC samples compared to normal tissue. Our data indicate that these genes may play a significant role in the pathogenesis of LSCC tumors since further functional investigation with three of them (HOXC8, HOXD10, and HOXD11) showed their involvement in biological processes related to tumor development as colony formation ability and cell migration. The results presented here support the hypothesis that aberrant expression of HOX genes is associated with larynx SCC development, and also warrant further investigation on the activity of HOX gene family in LSCC and their potential role as therapeutic targets.
References
IARC – International Agency for Research on Cancer. Globocan 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. https://www.iarc.fr. Accessed in 21 Jul 2015.
Hardisson D. Molecular pathogenesis of head and neck squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2003;260(9):502–8.
Zhang SY, ZM L, Luo XN, Chen LS, Ge PJ, Song XH, et al. Retrospective analysis of prognostic factors in 205 patients with laryngeal squamous cell carcinoma who underwent surgical treatment. PLoS One. 2013;8(4):e60157.
Silveira NJ, Varuzza L, Machado-Lima A, Lauretto MS, Pinheiro DG, Rodrigues RV, Severino P, Nobrega FG, Head and Neck Genome Project GENCAPO, Silva Jr WA, de B Pereira CA, Tajara EH. Searching for molecular markers in head and neck squamous cell carcinomas (HNSCC) by statistical and bioinformatic analysis of larynx-derived SAGE libraries. BMC Med Genet. 2008;11(1):56.
Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012;122(6):1951–7.
Colombo J, Fachel AA, De Freitas Calmon M, Cury PM, Fukuyama EE, Tajara EH, Cordeiro JA, Verjovski-Almeida S, Reis EM, Rahal P. Gene expression profiling reveals molecular marker candidates of laryngeal squamous cell carcinoma. Oncol Rep. 2009;21(3):649–63.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris III HA, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Petrelli F, Borgonovo K, Cabiddu M, Lonati V, Barni S. Relationship between skin rash and outcome in non-small-cell lung cancer patients treated with anti-EGFR tyrosine kinase inhibitors: a literature-based meta-analysis of 24 trials. Lung Cancer. 2012;78(1):8–15.
Cai J, Ma H, Huang F, Zhu D, Bi J, Ke Y, Zhang T. Correlation of bevacizumab-induced hypertension and outcomes of metastatic colorectal cancer patients treated with bevacizumab: a systematic review and meta-analysis. World J Surg Oncol. 2013;28:11–306.
Gore L, DeGregori J, Porter CC. Targeting developmental pathways in children with cancer: what price success? Lancet Oncol. 2013;14(2):e70–8.
Plaça JR, Bueno RBL, Pinheiro DG, Panepucci RA, Araújo LF, Mamede RCM, Figueiredo DLA, Silva Jr WA. Gene expression analysis of laryngeal squamous cell carcinoma. Genomics Data. 2015;(5):9–12.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot T, Malta TM, Pagnotta SM, Castiglioni I, Ceccarelli M, Bontempi G, Noushmehr H. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;5(8):44–e71.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25(4):402–8.
Broad Institute TCGA Genome Data Analysis Center. Correlations between copy number and mRNAseq expression. Broad Institute of MIT and Harvard. 2016. doi:10.7908/C19886DH.
Broad Institute TCGA Genome Data Analysis Center. Correlation between mRNA expression and DNA methylation. Broad Institute of MIT and Harvard. 2016. doi:10.7908/C15M653H.
Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–71.
Miyamoto K, Fukutomi T, Akashi-Tanaka S, et al. Identification of 20 genes aberrantly methylated in human breast cancers. Int J Cancer. 2005;116:407–14.
Cai LY, Abe M, Izumi S, et al. Identification of PRTFDC1 silencing and aberrant promoter methylation of GPR150, ITGA8 and HOXD11 in ovarian cancers. Life Sci. 2007;80:1458–65.
Furuta J, Nobeyama Y, Umebayashi Y, et al. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006;66:6080–6.
Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med (Berl). 2014;92:811–23.
Wang L, Chen S, Xue M, Zhong J, Wang X, Gan L, Lam EK, Liu X, Zhang J, Zhou T, Yu J, Jin H, Si J. Homeobox D10 gene, a candidate tumor suppressor, is downregulated through promoter hypermethylation and associated with gastric carcinogenesis. Mol Med. 2012;9(18):389–400.
Vardhini NV, Rao PJ, Murthy PB, Sudhakar G. HOXD10 expression in human breast cancer. Tumor Biol. 2014;35(11):10855–60.
Sekar P, Bharti JN, Nigam JS, Sharma A, Soni PB. Evaluation of p53, HoxD10 and E-cadherin status in breast cancer and correlation with histological grade and other prognostic factors. J Oncol. 2014;2014:7025–7.
Osborne J, Hu C, Hawley C, Underwood LJ, O’Brien TJ, Baker VV. Expression of HOXD10 gene in normal endometrium and endometrial adenocarcinoma. J Soc Gynecol Investig. 1998;5(5):277–80.
Li Q, Ding C, Chen C, Zhang Z, Xiao H, Xie F, Lei L, Chen Y, Mao B, Jiang M, Li J, Wang D, Wang G. miR-22 promotion of cell migration and invasion by targeting homeobox D10 gene in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2014;29(4):835–42.
Sharpe DJ, Orr KS, Moran M, White SJ, McQuaid S, Lappin TR, Thompson A, JA J. POUF2F1 activity regulates HOXD10 and HOXD11 promoting a proliferative and invasive phenotype in head and neck cancer. Oncotarget. 2014;5(18):8803–15.
Rodini CO, Xavier FC, Paiva KB, De Souza Setúbal Destro MF, Moyses RA, Michaluarte P, Carvalho MB, Fukuyama EE, Head and Neck Project Gencapo, Tajara EH, Okamoto OK, Nunes FD. Homeobox gene expression profile indicates HOXA5 as a candidate prognostic marker in oral squamous cell carcinoma. Int J Oncol. 2012;40(4):1180–8.
Adwan H, Zhivkova-Galunska M, Georges R, Eyol E, Kleeff J, Giese NA, Friess H, Bergmann F, Berger MR. Expression of HOXC8 is inversely related to the progression and metastasis of pancreatic ductal adenocarcinoma. Br J Cancer. 2011;105(2):288–95.
YB D, Dong B, Shen LY, Yan WP, Dai L, Xiong HC, Liang Z, Kang XZ, Qin B, Chen KN. The survival predictive significance of HOXC6 and HOXC8 in esophageal squamous cell carcinoma. J Surg Res. 2014;188(2):442–50.
Li Y, Chao F, Huang B, Liu D, Kim J, Huang S. HOXC8 promotes breast tumorigenesis by transcriptionally facilitating cadherin-11 expression. Oncotarget. 2014;5(9):2596–607.
Acknowledgments
We thank Amélia Goes de Araújo and Patrícia Vianna Bonini Palma for the technical support at Flow Cytometry Core at National Institute of Science and Technology in Stem Cell and Cell Therapy for the technical support. We also thank Josane de Freitas Sousa for the critical review of the manuscript final version. This work was supported by grant no. 140427/2010-4, National Counsel of Technological and Scientific Development (CNPq); grant no. 559809/2009-3 CNPq/GENOPROT; and grant nos. 2012/00588-5 and #2013/08135-2, São Paulo Research Foundation (FAPESP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Ribeirão Preto Medical School, University of São Paulo (USP) (Proc. No. 9371/2003). All patients underwent surgical resection at the Head and Neck Surgery Division of the Department of Ophthalmology, Otorhinolaryngology and Head & Neck of Ribeirão Preto Medical School, USP, and an informed consent was obtained from patients before surgery.
Conflicts of interest
None.
Additional information
Rafaela de Barros e Lima Bueno and Anelisa Ramão contributed equally to this work.
Electronic supplementary material
ESM 1
(DOCX 32879 kb)
Rights and permissions
About this article

Cite this article
de Barros e Lima Bueno, R., Ramão, A., Pinheiro, D.G. et al. HOX genes: potential candidates for the progression of laryngeal squamous cell carcinoma. Tumor Biol. 37, 15087–15096 (2016). https://doi.org/10.1007/s13277-016-5356-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5356-8