
Abstract
Armillaridin (AM) is an aromatic ester compound isolated from Armillaria mellea. Treatment with AM markedly reduced the viability of human chronic myelogenous leukemia K562, chronic erythroleukemia HEL 92.1.7, and acute monoblastic leukemia U937 cells, but not normal human monocytes, in a dose- and time-dependent manner. Treatment of K562 cells with AM caused changes characteristic of autophagy. Only a small amount of AM-treated K562 cells exhibited apoptosis. By contrast, AM treatment resulted in extensive apoptotic features in U937 and HEL 92.1.7 cells without evident autophagy. The autophagy of K562 cells induced by AM involved autophagic flux, including autophagosome induction, the processing of autophagosome-lysosome fusion and downregulation of BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3). By bcr-abl knockdown, the growth inhibition of K562 cells caused by AM was partially blocked, suggesting that AM-induced cell death might be a bcr-abl-dependent mode of autophagy-associated cell death. In conclusion, AM is capable of inhibiting growth and inducing autophagy-associated cell death in K562 cells, but not in normal monocytes. It may have potential to be developed as a novel therapeutic agent against leukemia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myelogenous leukemia (CML) is a disease of hematopoietic stem cells, arising from t(9;22)(q34;q11) translocation, resulting in the Philadelphia chromosome. This translocation leads to the juxtaposition of the abl gene from chromosome 9 and the bcr gene from chromosome 22, resulting in bcr-abl fusion genes that encode for bcr-abl transcripts and fusion proteins with unusual tyrosine kinase activities [1]. Signal transduction inhibitor-571 (STI-571), which can inhibit Bcr-abl tyrosine kinase activity, was first introduced in 1996 and then became the first-line therapy for CML, and it is regarded as a model for development of small-molecule targeted therapeutics. However, treatment of CML is changing rapidly because the primary curative treatment, allogeneic transplantation, has significant transplantation-related mortality; the target treatment has a better outcome, but 10–15 % of patients remain resistant to the tyrosine kinase inhibitor [2]. A number of pharmacological modalities have been tested for their ability to overcome imatinib-refractory CML, such as targeting another prosurvival pathway, autophagy [3]. Development of novel therapeutics against CML via distinct mechanisms is a legitimate strategy to overcome the current clinical dilemma.
Armillaria mellea is a medicinal fungus with a symbiotic relationship with the Chinese medicinal herb Gastrodia elata (tien-ma). It is commonly used to treat dizziness, headache, neurothenia, insomnia, numbness in limbs, and infantile convulsions. The extracts of A. mellea were reported to have antioxidant properties [4]. Armillaridin (AM), a sesquiterpenoid aromatic ester, is a compound isolated from the mycelium of A. mellea. It is a colorless, acicular compound with a boiling point of 132–134 °C [5]. The chemical structure is shown in Fig. 1. Although the antibacterial activities of AM have been reported, the biological properties of AM are not well understood. In our previous study, AM was reported to have therapeutic and radiosensitization effects on esophageal cancer cells [6].

Molecular structure of armillaridin (AM). AM is a sesquiterpenoid aromatic ester with antibacterial activity, C24H29O6Cl
Autophagy is characterized by the degradation of intracellular materials within lysosomes or vacuoles [7]. The process occurs at a basal level and in response to stress. Basal level autophagy plays a role in protein and organelle turnover and is rapidly upregulated when cells need to generate intracellular nutrients and energy. Autophagy and apoptosis are important to control the turnover of organelles and proteins in cells. Autophagy blocks the induction of apoptosis in general and apoptosis-associated caspase activation turns off the autophagic process. However, in some circumstances, autophagy may help to induce apoptosis or necrosis and lead to autophagic cell death [8].
In the present study, we demonstrated that AM inhibited the viability of various human leukemia cell lines and specifically induced autophagy-associated cell death in CML K562 cells in association with bcr-abl dependence and the downregulation of BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) expression.
Material and methods
Cells and reagents
Human leukemic cell lines, including the CML cell line K562 and two acute myelogenous leukemia cell lines, U937 (M5) and HEL92.1.7 (M6), were obtained from the American Type Culture Collection and were provided by Dr. Sheng-Yuan Wang at Taipei Veterans General Hospital, Taipei, Taiwan. They were kept in an RPMI-1640 medium (Gibco, NY, USA) with 10 % fetal bovine serum (FBS) (Biological Industries Ltd., Kibbutz Beit Haemek, Israel) and maintained in exponential growth conditions. HEL 92.1.7 cells were cultured in an RPMI-1640 medium with 10 % FBS and 1 % sodium pyruvate (Gibco, NY, USA); other leukemic cells were cultured in an RPMI-1640 medium with 10 % FBS. With approval of the IRB (No. 09MMHIS027) at MacKay Memorial Hospital, Taipei, Taiwan, human peripheral blood mononuclear cells were acquired from healthy volunteers using the Ficoll-Paque density gradient centrifugation (GE Healthcare, Buckinghamshire, UK). CD14+ cells were purified by high-gradient magnetic sorting using the miniMACS system (MiltenyiBiotec, BergischBladbach, Germany) with anti-CD14 microbeads. The purity of CD14+ monocytes was >95 % as determined by flow cytometry. Cells were kept in an αMEM medium (Gibco, NY, USA) with 10 % FBS for all experiments.
Armillaridin was provided by Dr. Chien-Chih Chen (Hungkuang University, Taichung, Taiwan). It was dissolved in DMSO as stock solutions with concentrations of 1.25, 2.5, 5, and 10 mM and then stored at 4 °C until use.
Chloroquine (Sigma-Aldrich, MO, USA) was dissolved in phosphate-buffered saline and stored at 4 °C. 3-Methyladenine (Sigma Aldrich, MO, USA) was dissolved into RPMI-1640. CQ was used at 10 μM, and 3-MA was used at 10 mM. The cells were preincubated with CQ and 3-MA for 90 min.
Cytoplasmic membrane integrity
The trypan blue exclusion assay was used to measure cell viability [9]. The numbers of viable cells were counted after 24, 48, and 72 h of exposure to AM.
Colony formation assay
Viable tumor cells (103) were plated into each 35-mm culture dish and allowed to grow in McCoy’s 5A medium containing 20 % heat-inactivated FCS and 0.24 % agarose at 37 °C in a humidified 5 % CO2 incubator. After 10 to 14 days, the dishes were stained with 0.4 % crystal violet and colonies containing ≥50 cells were counted.
Transmission electron microscopy
Cells were harvested, washed, and then fixed with cold 2.5 % glutaraldehyde in a 0.11 M cacodylate buffer for 30 min. Samples were then fixed in osmium tetroxide (1 %) and embedded in Epon resin (Electron Microscopy Sciences, PA, USA). Semi-thin sections were cut, stained with 5 % uranylacetate and lead citrate, and observed with a transmission electron microscope (JEM-1200EXII, JEOL, Co., Tokyo, Japan).
Cell cycle analysis by flow cytometry
After treatment, cells were washed with 10 % FBS/PBS and then fixed with ice-cold 70 % alcohol for 30 min. Cells were then incubated with Triton X-100 (Sigma-Aldrich, MO, USA), 0.2 mg/mL RNase A (Sigma-Aldrich, MO, USA), and 10 mg/mL propidium iodide (Sigma-Aldrich, MO, USA) for 10 min at 37 °C in the dark. Data were acquired using a FACSCalibur flow cytometer (Becton Dickinson, NJ, USA) and were analyzed by ModFIT software (Becton Dickinson, NJ, USA). A total of 104 cells were counted.
Assays for the detection of apoptotic events
Annexin V/PI staining (Trevigen Inc., MD, USA) was done according to the protocol provided by the manufacturer. Data acquisition and cell cycle analysis were performed on a FACSCalibur flow cytometer. STI-571 1 μM (Novartis Pharmaceutical Co., Switzerland) was used as a positive control for apoptosis induction in K562 cells [10]. Camptothecin 1 μM was used as a positive control for apoptosis induction in U937 cells [11] and HEL 92.1.0 cells.
Activity assay for caspases 3, 8, and 9
Caspase assays were also done according to the protocol provided by the manufacturer (Promega, WI, USA). The measurement of luminescence was done by luminometer.
GFP-LC3 assay
The green fluorescent protein (GFP) tagged-LC3 expressing vector was constructed by subcloning the LC3 coding sequence from pGST-LC3 plasmid (Provided by Dr. Tamotsu Yoshimori) into a pEGFP-C1 vector (Clontech, CA, USA). Cells were transiently transfected with GFP-LC3 by Lipofectamine™ 2000 (Invitrogen, CA, USA) based on the manufacturer’s protocol. After treatments with AM, cells were observed under a fluorescence microscope.
Endogenous LC3 and LAMP2A assay
K562 cells were fixed with paraformaldehyde and permeabilized with Triton X-100. Cells were then incubated with anti-LC3B antibody (Enzo Life Science, NY, USA) and anti-LAMP2A antibody (ABcam, MA, USA) in a press-to-seal silicone isolator. Then, cells were incubated with FITC-conjugated secondary anti-mouse antibody and Rhodamine-conjugated anti-rabbit secondary antibody. The nuclei of K562 cells were stained with DAPI. Cells were then examined under a fluorescence microscope.
shRNA for BCR knockdown
All lentivirus-based short hairpin RNA (shRNA) expression systems were obtained from the National RNAi Core Facility, Academia Sinica, Taiwan. LacO-inducible lentiviral vector pTRC-905-shBCR791 and 793 plasmids were constructed by the ligation of EcoRI- and AgeI-digested pTRC-905 vectors (a gift from the National RNAi Core Facility, Academia Sinica, Taiwan) with annealed oligonucleotides containing the same shRNA sequence against the BCR gene from pLKO.1-shBCR constructions (TCN0000000791 and TRCN0000000793). pLKO.1-shRFP was used to knockdown gene expression of was used to knock down gene expression of as non-targeting shRNA control. Lentiviruses were produced from 293T cells transfected with the vector controls pTRC-905-shBCR791, pTRC-905-shBCR793, or pLKO.1-shRFP plasmids, following the standard procedures from the National RNAi Core Facility [12]. After lentiviral infection and puromycin selection, K562 cells were incubated with or without 200 μM IPTG for 1 day and treated with DMSO, 5 or 10 μM AM for another 2 days. Flow cytometry was used to estimate the number of cells in the sub-G1 apoptotic phase, and the knockdown effects were estimated by monitoring the remaining amount of BCR protein expression.
Western blotting
Cells were cultured in 10-cm dishes in the presence or absence of AM at different concentrations. Those were lysed, and protein concentrations were determined by a bicinchoninic acid assay kit (Pierce, IL, USA). Equal amounts (50 mg in each lane) of protein extracts were electrophoresed on 10 % (w/v) SDS-polyacrylamide gels and then transferred onto nitrocellulose membranes. The membranes were incubated overnight at 4 °C with primary antibodies against LC3B (#2775), Phospho-p70S6K(#9205), p70S6K (#9202), Beclin-1(#4122), BCl-2 (#2876), c-ABL (#2862) (Cell Signaling Technology Inc., MA, USA), ATG5 (# PM050) (MBL international Cooperation, Japan), and BNIP-3 antibodies (ab10433) (ABcam, MA, USA) and then conjugated with horseradish peroxidase. The antigen-antibody reaction was detected under an enhanced chemiluminescence system (Abfrontier, Seoul, Korea) and then exposed to film. Quantification of the western blot analysis was done by ImageJ [13].
Data analysis
The experiments were performed in triplets. Results are reported as mean ± standard deviation. The Student’s t test and one-way analysis of variance followed by Scheffe’s test were used to estimate differences in the effects of AM on the viability of cells; the necrotic and apoptotic cells were tested by annexin V-PI staining. A p value less than 0.05 considered significant.
Results
Armillaridin reduced the viability of leukemic cell lines but not normal monocytes
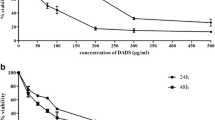
The viabilities of leukemic cells and CD 14+ monocytes were estimated. As shown in Fig. 2, AM inhibited the viability of leukemic cell lines K562, U937, and HEL 92.1.7 in a dose- and time-dependent manner. The estimated IC50 values at 48 h were 4.4 μM for K562, 3.0 μM for U937, and 3.7 μM for HEL 92.1.7. It was indicated that the clonogenic capacities of the cells have been changed by colony formation assay (Table 1). In contrast, AM had no significant effect on the viability of CD14+ monocytes (IC50 > 10 μM). The results indicate that AM might preferentially inhibit the viability of leukemic cells.

AM reduced the viability of human leukemic cell lines and the normal counterpart, CD 14+ monocytes. All cells were treated with various concentrations of AM for 24, 48, or 72 h. Trypan blue exclusion tests were performed to examine cell viability. Data from three separate experiments are expressed as mean ± standard deviation. *p < 0.05. a K562 cells, b U937 cells, c HEL 92.1.7 cells, d CD14+ monocytes
Morphological changes after AM treatment
As shown in Fig. 3a, the AM-treated K562 cells showed various cytoplasmic vacuoles with intact cell membranes and intact nuclei characteristic of autophagy. A small number of cells with apoptotic features, including reduced cell volume, chromatin condensation, and nuclear fragmentation, were also found. However, as demonstrated in Fig. 3b, c, AM treatment resulted in extensive apoptotic features in U937 and HEL 92.1.7 cells without evident autophagy.

Morphological changes of leukemic cells under AM treatment. Cells were treated with 10 μM AM for 24 or 48 h. Light microscopy at magnification ×1000 was used for morphological observation. a K562 cells, b U937 cells, c HEL 92.1.7 cells
Effect of AM on recruitment of LC3 to autophagosomes in K562 cells
Fluorescent microscopy of starved cells treated with AM revealed a punctate pattern of GFP-LC3 aggregation, indicating recruitment of LC3 to autophagosomes (Fig. 4, starvation). To examine whether the autophagosomes were efficiently delivered to lysosomes in AM-exposed cells, we performed immunofluorescence for both LC3B and lysosome-associated membrane protein type 2A (LAMP2A), a receptor in the lysosome membrane. The punctate of LC3B and LAMP2A merged together in both AM-exposed and starvation-triggered cells. It indicates that the process induced by AM is autophagy induction rather than autophagosome accumulation (Fig. 4). The ultrastructural morphological changes were examined by transmission electron microscopy in K562 cells as shown in Fig. 5a. There were various vesicles measuring approximately 1 μm in diameter within the cytoplasm, and the nucleus and cell membrane were intact. Fusion vesicles and the remnants of organelles in the vesicles were also noted. Annexin V staining revealed that the exposure of cells to imatinib (1 μM) alone for 48 h resulted in marked apoptotic events [14]. No obvious apoptotic or necrotic events were noted in AM-treated K562 cells (Fig. 5b). To compare the amount of apoptotic population in each cell line, we performed Annexin V/PI flow cytometry analysis and found the proportion of early apoptotic population (Annexin V positive and PI negative) in K562, U937, and HEL 92.1.7 cells by AM 10 μM treatment for 48 h was 1.86 ± 0.18, 2.69 ± 0.18, and 5.88 ± 2.32 % and the late apoptotic and necrotic population (Annexin V positive and PI positive) in K562, U937, and HEL 92.1.7 cells by AM 10 μM treatment for 48 h was 4.51 ± 2.09, 7.94 ± 1.24, and 33.03 ± 1.64 %, respectively. We also examined the enzymatic activity of caspase 3/7, caspase 8, and caspase 9. AM did not activate these caspases. By contrast, the STI-571 activated these caspases during the condition-induced apoptosis of K562 cells (Table 2).

AM induced autophagy in leukemia cells. Plasmid carrying GFP-LC3 was transfected into K562 cells prior to treatment. Punctate pattern aggregation of GFP-LC3, indicating recruitment of LC3 to autophagosome, was observed by fluorescent microscopy in starved cells treated with AM. Three independent tests were done. Endogenous LC3B and LAMP2A were observed by fluorescent microscopy in cells treated with AM and starvation. Magnification ×1000. Blue, DAPI. Green, FITC LC3B. Red, Rhodamine-LAMP2A

AM induced autophagy without evident apoptosis in chronic myelogenous leukemia cells. a Transmission electron microscopy at magnification ×8000–25,000 was used for the morphological observation of AM-treated K562 cells. b Cells were treated with 0.1 % DMSO, 5 μM AM, or 1 μM STI-571 (as a positive control of apoptosis) for 48 h. Harvested cells were stained with propidium iodide for necrotic cells and Annexin V for externalization of phosphatidylserine. Flow cytometry was used for analysis. Data from three separate experiments are expressed as mean ± standard deviation. *p < 0.05
Autophagic flux in AM-treated K562 cells
Concentration-dependent LC3-II blotting revealed that LC3-II expression increased in a concentration-dependent manner by treatment with AM in K562 cells (Fig. 6a). The cells were incubated for a longer time, to 72 h. It showed that LC3-II accumulated at 48 h and disappeared at 72 h after treatment, indicating an active autophagic flux rather than a persistent accumulation of autophagosomes (Fig. 6b). The accumulation of LC3-II was also found in AM-treated U937 and HEL 92.1.0 cells (Fig. 6c).

Effect of AM on expression of LC3B in K562 cells. a K562 cells were treated with serial concentrations of AM for 24 h. Protein extracts were subjected to western blotting. Data from three separate experiments showed a similar trend. Lane 1, untreated; lane 2, 0.1 % DMSO; lane 3, 1.25 μM AM; lane 4, 2.5 μM AM; lane 5, 5 μM AM; lane 6, 10 μM AM; lane 7, starvation for 6 h. b K562 cells were treated with 10 μM of AM for 24, 48, and 72 h. Protein extracts were subjected to western blotting. Data from three separate experiments showed a similar trend. Lane 1, untreated; lane 2, 0.1 % DMSO; lane 3, AM 24 h; lane 4, AM 48 h; lane 5, AM 72 h; lane 6, starvation 6 h. c U937 and HEL 92.1.7 cells were treated with 10 μM AM for 24 h. Protein extracts were subjected to western blotting. Lane 1, untreated; lane 2, 0.1 % DMSO; lane 3, 10 μM AM; lane 4, post 6-h starvation
We assessed autophagic flux by combining chloroquine (CQ) or 3-methyladenine (3-MA) with AM and found that pretreatment with 10 μM of CQ enhanced the accumulation of LC3-II in K562 cells, as shown in Fig. 7. In contrast, pretreatment with 10 mM of 3-MA resulted in a decreased accumulation, indicating that AM-induced autophagy affected autophagic flux through the induction to autophagosome-lysosome fusion. There was an increased survival with pretreatment of CQ (50.85 vs. 65.17 %) and a decreased survival with pretreatment of 3-MA (50.85 vs. 35.15 %), according to colony formation assay (Table 1).

Effect of AM on expression of LC3B under autophagy inhibitor in K562 cells. Cells were treated with indicated conditions of AM for 24 h. Protein extracts from whole cells were subjected to western blotting. Lane 1, 0.1 % DMSO; lane 2, 10 μM AM; lane 3, pretreated with 10 μM chlorquine (CQ); lane 4, pretreated with 10 μM CQ and then treated with 10 μM AM; lane 5, pretreatment of 10 mM 3-methyladenine (3-MA); lane 6, pretreatment of 10 mM 3-MA and then treated with 10 μM AM. Mean band intensities of LC3B-II are shown in a and b
Mechanisms of AM-induced autophagy
Key molecules related to autophagy were expressed in a time-dependent manner (Fig. 8). Exposure to AM resulted in a time-dependent decrease in BNIP3 expression in K562 cells. The expression of phosphorylated p70s6k showed an initial increase followed by a decline during 24-h treatment. The expression of p70s6k, Atg5, and beclin-1 showed no evident changes.

Autophagy-related signaling in AM-treated K562 cells. Cells were treated with 10 μM of AM for indicated times. Protein extracts from whole cells were subjected to western blotting. Data from three separate experiments showed a similar trend. Lane 1, 0 h; lane 2, 2 h; lane 3, 4 h; lane 4, 8 h; lane 5, 16 h; lane 6, 24 h; lane 7 starvation for 6 h. Mean band intensity of LC3B-II compared with starvation group is shown
Blockage of AM-induced cytotoxicity in BCR-ABL knockdown K562 cells
The main difference between K562 cells and acute myelogenous leukemic cells are the bcr-abl fusion gene. Knockdown of bcr-abl was performed with an isopropyl-β-d-thio-galactoside (IPTG) induction system and was validated by western blotting analysis (Fig. 9a). The bcr-abl knockdown partially blocked AM-induced cytotoxicity and apoptosis in K562 cells (Figs. 9b, c), indicating that AM-induced autophagy might be mediated by a BCR-ABL-dependent mode of autophagy-associated cell death.

Effect of shBCR on cell growth inhibition in K562 cells. K562 cells were infected with lentivirus produced from pLKO.1-shRFP (shRFP), pTRC-905-shBCR791 (shBCR791), or pTRC-905-shBCR793 (shBCR793). After being selected with puronycin, cells were incubated with or without 200 μM of IPTG for the induction of shRNA expression for 1 day, and then treated with DMSO, 5 or 10 μM of AM for 2 days. The knockdown effect of the bcr-abl gene was detected by western blot analysis using antibodies against bcr and actin (a). Data from three separate experiments showed a similar trend. Growth inhibition effects (b) and percent of apoptotic cells (c) under AM treatment were detected by MTT assay and flow cytometry
Discussion
The results of this study showed that AM reduced cell viability and induced extensive autophagy in human leukemic cells. Current data demonstrate that AM affects autophagy flux in LC3 turnover assays. This autophagic event involved down-regulation of BNIP3 and was independent of the early externalization of phosphatidylserine, an early event of apoptosis. It also showed that AM preferentially inhibited the viability of myeloid leukemic cells without marked toxicity to the normal counterpart monocytes.
AM treatment caused the downregulation of BNIP3 expression in CML K562 cells. The upregulation of BNIP3, a proapoptotic protein that binds to antiapoptotic proteins of the Bcl-2 family, may cause autophagy in hypoxia-induced autophagy [15]. BNIP3 is also a proautophagic protein and induces autophagy by disrupting the Bcl-2-beclin-1 interaction and activating beclin-1 [16]. The upregulation of BNIP3 in autophagy was revealed in ceramide-induced and arsenic-trioxide-induced autophagic cell death in malignant glioma cells and in platonin-induced autophagic cell death in leukemia cells in prior studies [17–19]. There were no obvious changes in bcl-2 and beclin-1 in K562 cells treated with AM. It must also be mentioned that loss of BNIP3 was protective against hypoxia-induced autophagic cell death [15]. Conversely, in our study, the decreased expression of BNIP3 by AM was evident in a time-dependent manner. Given that BNIP3 is known as tumor suppressor and contributor to hypoxia-induced autophagy, further elucidation of the role of BNIP3 in AM-induced autophagy in K562 cells is needed.
Our results showed relatively higher survival rates by pretreating with the lysosomotropic reagent CQ before administering AM, which indicated that AM induced autophagy-associated cell death through the autophagy process. Pretreatment of 3-MA at the condition mitigating the LC3-II conversion (as shown in Fig. 5c) further reduced the survival of AM-treated K562 cells. The role of 3-MA may counteract the induction of autophagy with accompany by promoting apoptosis. It implicates that the development of autophagy and apoptosis seems an interchangeable or interactive process. For example, apoptosis induced by calpain inhibitor decreased oridonin-induced autophagy. By contrast, the inhibition of autophagy by 3-MA increased oridonin-induced apoptosis in L929 cells [20].
We showed that AM induced autophagy-associated cell death in K562 cells and apoptosis in a small population of K562 cells, whereas AM induced extensive apoptosis and autophagy in U937 and HEL 92.1.7 cells. The opposite roles played by autophagy are not only dependent on the cell type but, more importantly, on the entity or duration of stress conditions. We also found that bcr-abl knockdown partially blocked AM-induced cytotoxicity and apoptosis in K562 cells. It is known that the bcr-abl fusion gene exists in K562 cells but not in other leukemic cell lines we used, suggesting that the bcr-abl fusion gene might play a role in triggering autophagy-associated cell death rather than apoptotic cell death.
Gousseties et al. [21] demonstrated “that decreasing BCR-ABL expression by arsenic trioxide lead autophagy in K562 cells involving p62/SQSTM1-medicated localization of the oncoprotein to the autolysosomes and subsequent degradation medicated by the protease cathepsin B.” This event could be reversible by inhibition of autophagy. Bellodi et al. [3] showed that the inhibition of BCR-ABL with STI-571 induced apoptosis and autophagy, and autophagy inhibits apoptosis in CML cells. Sheng et al. [22] demonstrated that BCR-ABL suppressed autophagy through the regulation of mTOR transcription. The interplay of BCR-ABL and autophagy varies depending on the different circumstances. In our study, AM induced autophagy-associated cell death without involving BCR-ABL expression (Supplemental Fig. 1 ). Knockdown of bcr-abl resulted in less growth inhibition and a greater hypoploidy population in K562 cells treated with AM. It suggests that the development of autophagy in AM-treated K562 cells might be counteracted in a bcr-abl-dependent manner.
Conclusion
In summary, armillaridin is capable of inhibiting growth in human leukemia cells but not in normal monocytes. Armillaridin-induced cell death might be a bcr-abl-dependent mode of autophagy-associated cell death in CML K562 cells. It could be considered a novel therapeutic drug against leukemia.
Abbreviations
- AM:
-
Armillaridin
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa interacting protein 3
- CML:
-
Chronic myelogenous leukemia
- CQ:
-
Chloroquine
- 3-MA:
-
3-Methyladenine
- STI-571:
-
Signal transduction inhibitor-571
References
Hehlmann R, Hochhaus A, Baccarani M, European L. Chronic myeloid leukaemia. Lancet. 2007;370:342–50.
Apperley JF. Chronic myeloid leukaemia. Lancet. 2014.
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, Dinsdale D, Tirro E, Vigneri P, Nicotera P, Dyer MJ, Holyoake T, Salomoni P, Calabretta B. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23.
Lung MY, Chang YC. Antioxidant properties of the edible Basidiomycete Armillaria mellea in submerged cultures. Int J Mol Sci. 2011;12:6367–84.
Yang JS, Chen YW, Feng XZ, Yu DQ, Liang XT. Chemical constituents of Armillaria mellea mycelium. I. Isolation and characterization of armillarin and armillaridin. Planta Med. 1984;50:288–90.
Chi CW, Chen CC, Chen YJ. Therapeutic and radiosensitizing effects of armillaridin on human esophageal cancer cells. Evidence-based complementary and alternative medicine: eCAM. 2013;2013:459271.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94.
Strober W. Trypan blue exclusion test of cell viability. Current protocols in immunology/edited by John E Coligan [et al]. 2001;Appendix 3:Appendix 3B.
Puissant A, Grosso S, Jacquel A, Belhacene N, Colosetti P, Cassuto JP, Auberger P. Imatinib mesylate-resistant human chronic myelogenous leukemia cell lines exhibit high sensitivity to the phytoalexin resveratrol. FASEB J. 2008;22:1894–904.
Sen N, Das BB, Ganguly A, Mukherjee T, Tripathi G, Bandyopadhyay S, Rakshit S, Sen T, Majumder HK. Camptothecin induced mitochondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ. 2004;11:924–36.
Huang HL, Chen YC, Huang YC, Yang KC, Pan H, Shih SP, Chen YJ. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia K562 cells. PLoS One. 2011;6:e29014.
Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Jacquel A, Herrant M, Legros L, Belhacene N, Luciano F, Pages G, Hofman P, Auberger P. Imatinib induces mitochondria-dependent apoptosis of the Bcr-Abl-positive K562 cell line and its differentiation toward the erythroid lineage. FASEB J. 2003;17:2160–2.
Azad MB, Gibson SB. Role of BNIP3 in proliferation and hypoxia-induced autophagy: implications for personalized cancer therapies. Ann N Y Acad Sci. 2010;1210:8–16.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–93.
Chen YJ, Huang WP, Yang YC, Lin CP, Chen SH, Hsu ML, Tseng YJ, Shieh HR, Chen YY, Lee JJ. Platonin induces autophagy-associated cell death in human leukemia cells. Autophagy. 2009;5:173–83.
Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein bnip3. Oncogene. 2005;24:980–91.
Cheng Y, Qiu F, Huang J, Tashiro S, Onodera S, Ikejima T. Apoptosis-suppressing and autophagy-promoting effects of calpain on oridonin-induced L929 cell death. Arch Biochem Biophys. 2008;475:148–55.
Goussetis DJ, Gounaris E, EJ W, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120:3555–62.
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118:2840–8.
Acknowledgments
We want to thank Dr. King-Song Jeng and the National RNAi Core Facility, Academia Sinica, Taiwan, for shRNA knockdown techniques, the pTRC-905 vector, and the pTRC-905-shBCR constructions. This work was supported by grant NSC98-2323-B-241 from the National Science Council, Taiwan, and grant 09MMHIS027 and MMH-E-105-13, E-104-13 from MacKay Memorial Hospital, Taiwan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None.
Electronic supplementary material
Supplemental Figure 1.
Effect of armillaridin on BCR-ABL expression on K562 cells. Cells were untreated, treated with 0.1% DMSO (solvent), 10 μM armillaridin for 24 hours or starvation for 6 hours. Protein extracts from whole cells were subjected to Western blotting. Data from three separate experiments showed similar trend. No significant differences on the expression of BCR-ABL were found between control and armillaridin treatment. (GIF 175 kb)
Rights and permissions
About this article

Cite this article
Chang, WH., Huang, HL., Huang, WP. et al. Armillaridin induces autophagy-associated cell death in human chronic myelogenous leukemia K562 cells. Tumor Biol. 37, 14291–14300 (2016). https://doi.org/10.1007/s13277-016-5208-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5208-6