
Abstract
Long noncoding RNAs (lncRNAs) are fundamental regulators of pre- and post-transcriptional gene regulation. Over 35,000 different lncRNAs have been described with some of them being involved in cancer formation. The present study was initiated to describe differentially expressed lncRNAs in basal cell carcinoma (BCC). Patients with BCC (n = 6) were included in this study. Punch biopsies were harvested from the tumor center and nonlesional epidermal skin (NLES, control, n = 6). Microarray-based lncRNA and mRNA expression profiles were identified through screening for 30,586 lncRNAs and 26,109 protein-coding transcripts (mRNAs). The microarray data were validated by RT-PCR in a second set of BCC versus control samples. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of mRNAs were performed to assess biologically relevant pathways. A total of 1851 lncRNAs were identified as being significantly up-regulated, whereas 2165 lncRNAs were identified as being significantly down-regulated compared to nonlesional skin (p < 0.05). Oncogenic and/or epidermis-specific lncRNAs, such as CASC15 or ANRIL, were among the differentially expressed sequences. GO analysis showed that the highest enriched GO targeted by up-regulated transcripts was “extracellular matrix.” KEGG pathway analysis showed the highest enrichment scores in “Focal adhesion.” BCC showed a significantly altered lncRNA and mRNA expression profile. Dysregulation of previously described lncRNAs may play a role in the molecular pathogenesis of BCC and should be subject of further analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is widely recognized that almost 70–98 % of all transcriptional output in human cells consists of noncoding RNAs (ncRNAs), whereas only 1.9 % are actually transcribed into protein-coding mRNAs [1]. This expanding group of ncRNAs can be divided into two groups, small ncRNAs (sncRNAs) and long ncRNAs (lncRNAs). The most prominent members belonging to the group of sncRNAs are microRNAs (miRNAs), which represent pivotal regulators of protein expression on a post-transcriptional level and are 17–23 nt long, exerting both tumor suppressor and oncogenic effects that participate in basal cell carcinoma (BCC) formation, which is the most common form of skin cancer and human cancer in general [2–7]. Similar to miRNAs, lncRNAs are defined as nonprotein-coding RNAs with tissue-specific expression and have been shown to be decisive regulators by controlling gene expression on the pre- and post-transcriptional level [8]. In contrast to the well-studied group of sncRNAs, lncRNAs range in length from >200 nt up to 100 kb [9]. A vast number of studies have demonstrated their pivotal role in controlling tissue development and their strong impact on fundamental central cellular processes, such as epigenetic modifications of chromatin, gene regulation, nuclear import, and X chromosome inactivation, in both normal and disease tissue [10]. The initial presumption that ncRNA sequences are unnecessary transcriptional noise or accumulated transcriptional waste originating from evolutionary processes that were genetically carried from one evolutionary state to the next has been shown to be vastly untrue. LncRNAs have currently emerged as the largest group of molecules transcribed by human translational machinery. The number and diversity of lncRNAs massively exceeds the approximately 19,000 protein-coding human genes, and the characterization of their intracellular role is constantly being updated [11]. The tremendous biological diversity of lncRNAs hints at their various levels of regulation, which includes epigenetic, transcriptional, and post-transcriptional gene regulation, as well as regulation of sncRNAs, such as miRNAs [12]. One important characteristic is their ability to form ribonucleoprotein complexes that directly regulate gene expression in the nucleus. As previously mentioned, lncRNAs have a broad range of effects and have been shown to control cell cycle, proliferation, differentiation, and apoptosis; more functions are still being discovered [13]. The maintenance of stem cells (keratinocyte progenitor) and terminal differentiation of keratinocytes in the epidermis have been shown to be affected by a defined set of lncRNA genes [14–17]. Keratinocyte homeostasis depends on lncRNAs as they play key roles in differentiation. At the same time, disturbance of keratinocyte lncRNA expression can potentially result in cancer formation. This is very likely as there is growing evidence that lncRNAs are involved in malignancies and tumor development, in general [13, 18, 19]. Although most other forms of cancer, including cutaneous melanoma, have been successfully investigated regarding differential lncRNA expression, nonmelanoma skin cancers such as BCC, which affects keratinocytes, have not yet been systematically evaluated [10]. This study was performed to describe differentially expressed lncRNAs in BCC tissue.
Materials and methods
The procedures performed in this study were in accordance with the ethical standards of the institutional research committee and with the 1964 Declaration of Helsinki and its later amendments and comparable ethical standards. Informed consent was obtained from all individuals who were included in the study.
Samples
A total of 12 samples (6 BCC and 6 controls) from 12 individuals (5 women, 7 men, mean age 71.1 years) were enrolled in this study (Table 1). All individuals were of Caucasian origin and had no skin disease other than BCC. There is no statistical difference in age between BCC and control (p > 0.05). While excising BCCs with cold steel under local anesthesia, 4-mm punch biopsies were taken from the center of the tumor and from nonlesional epithelial skin (control). These samples were immediately placed in RNAlater (Qiagen, Hilden, Germany) and stored at −80 °C. Total RNA was extracted with TRIzol (Life Technologies, Carlsbad, USA) according to the manufacturer’s protocol. RNA quantity and quality were measured with a NanoDrop ND-1000 spectrophotometer, and RNA integrity was assessed by standard denaturing agarose gel electrophoresis. Two groups of samples, BCC_1, BCC_2, and BCC_3 and Control_1, Control_2, and Control_3, were used for microarray scanning of lncRNA and mRNA expression. The samples BCC_4, BCC_5, and BCC_6 and Control_4, Control_5, and Control_6 were used for qRT-PCR validation as a second independent group of samples.
Microarrays
Microarrays were performed with Arraystar Human LncRNA Microarray V3.0 (Rockville, Maryland, USA), which is designed for global profiling of human lncRNAs and protein-coding transcripts (mRNAs). The following target sequences were represented in the array: 30,586 lncRNAs, 26,109 coding transcripts (mRNAs), 709 lncRNAs containing an open reading frame and sharing the same start codon with protein-coding transcripts, and 577 transcribed pseudogenes. The following lncRNA sources were represented in the array: 3991 lncRNAs from Reference Sequence (RefSeq; http://www.ncbi.nlm.nih.gov/refseq/), 12,694 lncRNAs from the UCSC Known Genes dataset (Known Genes 6; http://genome.ucsc.edu/cgi-bin/hgTables), 20,596 lncRNAs from the GENCODE database of annotations for all human protein-coding and noncoding genes, 1492 lncRNAs from the RNAdb database (http://research.imb.uq.edu.au/rnadb/), 1112 lncRNAs from the Noncoding RNA Expression Database (NRED; http://jsm-research.imb.uq.edu.au/nred/cgi-bin/ncrnadb.pl), 287 lncRNAs from the lncRNAdb (http://lncrnadb.com/), 2592 lincRNAs described by Khalil et al., 14,353 transcripts expressed from 4662 stringently defined human lincRNA genes described by Cabili et al., 475 ultraconserved regions (UCRs) described by Bejerano et al. (http://users.soe.ucsc.edu/~jill/ultra.html), 407 transcribed regions within the four HOX loci in humans identified by Rinn et al., and 3019 human lncRNAs with enhancer-like functions described by Orom et al. [16, 20–28].
RNA labeling and array hybridization
Sample labeling and array hybridization were performed according to the Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technology, Santa Clara, USA) as previously described [29]. After removal of rRNA (mRNA-ONLY™ Eukaryotic mRNA Isolation Kit, Epicentre), mRNA was purified from total RNA. A mixture of oligo(dT) and random priming method (Arraystar Flash RNA Labeling Kit, Arraystar, Rockville, USA) was used to amplify each of the samples and transcribe them into fluorescent cRNA along the entire length of the transcripts. Labeled cRNAs were then purified by the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. A NanoDrop ND-1000 spectrophotometer was used to measure the concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA). Then, 5 μl of 10× blocking agent and 1 μl of 25× fragmentation buffer were added to 1 μg of each labeled cRNA, and the mixture was heated at 60 °C for 30 min. Next, 25 μl of 2× GE hybridization buffer was added to dilute the labeled cRNA. Finally, 50 μl of the hybridization solution was dispensed into the gasket slide and assembled onto the lncRNA expression microarray slide, which was incubated for 17 h at 65 °C in an Agilent Hybridization Oven (SureHyb Microarray Hybridization Chamber, Agilent Technologies, Santa Clara, USA). The hybridization arrays were washed, fixed, and scanned with an Agilent DNA Microarray Scanner G2505C (Agilent Technologies, Santa Clara, USA).
Quantitative real-time reverse transcription polymerase chain reaction
To validate the microarray data, the expression levels of the three up-regulated lncRNA genes, ENST00000510016, ENST00000560097, and ENST00000560054; the three down-regulated lncRNA genes, ENST00000504509, ENST00000523831, and ENST00000558730; and the reference gene, β actin, were determined in a second set of BCC (BCC_4, BCC_5, BCC_6) and control (Control_4, Control_5, Control_6) samples. Briefly, cDNA was synthesized; a standard curve was prepared, and gene expression was determined by real-time reverse transcription polymerase chain reaction (RT-PCR). The following components were mixed together for first-strand cDNA synthesis: 1 μl of 10 μM oligo(dT)18 primer, 0.3 μl of 10 μM random (N9) primer, 1.0 μg of total RNA, 1 μl of 10 mM dNTP Mix (Invitrogen, Waltham, USA), and sterile distilled water, for a total volume of 13 μl. The mixture was heated to 65 °C for 5 min, followed by incubation on ice for at least 1 min. The contents of the tube were mixed by pipetting gently up and down after adding the following components: 4 μl of 5× first-strand buffer (Invitrogen, Waltham, USA), 1 μl of 0.1 M dl-Dithiothreitol (DTT; Invitrogen, Waltham, USA), 1 μl of RNase Inhibitor (Enzymatics, Beverly, USA), and 1 μl of SuperScript III RT (Invitrogen, Waltham, USA). After incubation at 50 °C for 60 min, the reaction was inactivated by heating at 70 °C for 15 min (ViiA 7 Real-Time PCR System, Applied Biosystems, Carlsbad, USA). A total of 200 μl of ddH2O was added to every 20 μl of cDNA synthesis reaction. The finished first-strand cDNA synthesis reaction was maintained on ice until standard curve preparation. Then, 5 μl of the Arraystar PCR Master Mix (Arraystar Inc., Rockville, USA), 1 μl of 10 μM PCR forward primer, 1 μl of 10 μM PCR reverse primer, 1 μl of diluted first strand cDNA, and 2 μl of ddH2O were mixed together for a total volume of 10 μl. The following primer pairs were used: ENST00000510016 (F 5′-GTCAGCACCGCAGCAAATC-3′; R 5′-CATCACTCACCGCTGTTTTCA-3′), ENST00000560097 (F 5′-GAGTGACCCGATTTTCCAGC-3′;
R 5′-GGTCTCTCTGACCTTCTCCTTG-3′), ENST00000560054 (F 5′-GCATTAGCGTTTTTTATTGGAG-3′; R: 5′-AATGCCCTTTTAGGCTTGAT-3′), ENST00000504509 (F 5′-GAACAAGACCAGGAGGAGGTTT-3′; R 5′-GCCAATCATCGCTGTGAGGTA-3′), ENST00000523831 (F 5′-GTGTAGAGAGTGGTGTGGGGA-3′; R 5′-GCTTGGCTCATAGGTCAGGAT-3′), and ENST00000558730 (F 5′-ACACCCAACATCCACACCCTx-3′; R 5′-TGCAGATCCTCATCCCGTAA-3′). The 384-well PCR array plates were loaded, and RT-PCR was run in relative quantification mode with triplicate measurements. The following temperature profiles were used: polymerase activation/denaturation at 95 °C for 10 min, followed by 40 amplification cycles at 95 °C for 10 s and 60 °C for 1 min. To quantify the PCR products, a standard curve was constructed using serial tenfold dilutions (from 1 to 10−6). Finally, RT-PCR was prepared by mixing 5 μl of Arraystar PCR Master Mix, 0.6 μl of 10 μM PCR forward primer, 0.6 μl of 10 μM PCR reverse primer, 2 μl of diluted first-strand cDNA synthesis reaction, and 1.8 μl of ddH2O to a total volume of 10 μl. The following temperature profiles were used: polymerase activation/denaturation 95 °C for 10 min, followed by 40 amplification cycles at 95 °C for 10 s and 60 °C for 1 min. The gene concentration of each sample was generated by ABI7900 Analysis Software SDS2.3 (Applied Biosystems, Carlsbad, USA).
Bioinformatics data analysis
The data from this study were deposited in the Gene Expression Omnibus (GEO) of the National Center for Biotechnology Information and are accessible through GEO Series accession number GSE74858 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74858).
Array images were analyzed with the Agilent Feature Extraction software (version 11.0.1.1). The GeneSpring GX v12.0 software package (Agilent Technologies, Santa Clara, USA) was used for quantile normalization and subsequent data processing. Quantile normalization was applied to the raw data. In detail, the lncRNAs and mRNAs that had at least three out of six samples flagged in Marginal or Present (“All Targets Value”) were chosen for data analysis. Volcano plot filtering between two groups (BCC vs. control) was used to identify differentially expressed lncRNAs and mRNAs with statistical significance. R software (version 2.15, http://www.r-project.org/) was used for hierarchical clustering to arrange samples into groups based on their expression levels. Pathway analysis and Gene Ontology (GO) analysis were performed with the standard enrichment computation method.
GO and KEGG pathway analysis
GO analysis, which is a functional analysis associating differentially expressed mRNAs with GO categories derived from GO (http://www.geneontology.org), was performed as previously described [30]. GO composed of three structured networks of defined terms (cellular component (cc), biological process (bp), and molecular function (mf)) that describe gene product attributes has enabled a broader understanding of the microarray data [31]. This was made possible by grouping genes of interest into cc, bp, and mf categories. The significance of GO term enrichment in differentially expressed mRNAs was assessed by calculating enrichment scores with Fisher’s exact test, comparing a portion of the gene list in the BCC group to the control group, and p value and false discovery rates (FDR) using the method described by Benjamini and Hochberg [32]. Furthermore, a pathway analysis for differentially expressed mRNAs was performed based on the latest Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg) database to determine biological pathways for the significant enrichment of differentially expressed mRNAs with a possible impact on BCC tumor formation.
Statistical analysis
For statistical analysis of lncRNA and mRNA microarray expression data, an unpaired t test was performed after adjusting the original p values by applying the algorithm devised by Benjamini and Hochberg using MedCalc software version 15.2 (MedCalc, Mariakerke, Belgium) [32]. To identify differential lncRNA and mRNA expression between the BCC and control samples, fold changes (FC) were calculated and filtered. Statistical significance and robust detection were evaluated to identify differentially expressed lncRNA and mRNA in BCC. Statistical significance was defined as differential expression by a p ≤ 0.05 with an FC ≥ 2.0 and selected as differentially repressed by a p ≤ 0.05 with an FC ≥ −2.0. Using Microsoft Excel’s data sort and filter functionalities (Microsoft, Redmond, USA), analysis outputs were filtered and differentially expressed with lncRNAs and mRNAs ranked by FC and p value (p ≤ 0.05). Microarray data were visualized by the “heatmap.2” function in the “gplots” package that was used for heat map generation. Cluster analysis with Euclidean distance as a measure was performed as previously described [33, 34]. Correlation analysis was performed by calculating Pearson’s correlation coefficient r for each sample within the two groups and for all pairwise comparisons.
Results
Quality control
RNA quantity and quality were measured with a NanoDrop ND-1000 spectrophotometer, and RNA integrity was assessed by standard denaturing agarose gel electrophoresis after RNA extraction and prior to sample labeling. The inclusion criteria for RNA were an O.D. A260/280 ratio between 1.8 and 2.1 and an O.D. A260/230 ratio >1.8. RNA integrity was successfully tested by denaturing agarose gel electrophoresis. The 28S and 18S ribosomal RNA bands were sharp and intense. The intensity of the upper band, which was approximately twice that of the lower band, and the absence of a high-molecular-weight smear or band above the 28S ribosomal RNA indicated that there was no DNA contamination. RT-PCR showed that the three up-regulated lncRNAs were up-regulated and that the three down-regulated lncRNAs were down-regulated, validating microarray data in a second independent set of BCC versus control samples. The following ratios for gene expression were calculated (BCC/control) for the up-regulated genes, ENST00000510016 (2.35), ENST00000560097 (276.07), and ENST00000560054 (4.08), and the down-regulated genes, ENST00000504509 (0.03), ENST00000523831 (0.32), and ENST00000558730 (0.01).
Identification of differentially expressed lncRNAs and mRNAs in BCC
A total of 1851 lncRNAs were significantly up-regulated, and 2165 lncRNAs were significantly down-regulated with the criteria of FC ≥ 2 and p < 0.05 (Table 2). The most up-regulated lncRNA was AC073135.3 (FC 183.72), and the most down-regulated was RP11-350F16.1 (FC 98.75). Using the same criteria, FC ≥ 2 and p < 0.05, 1477 mRNAs were up-regulated and 1567 mRNAs down-regulated (Table 3). The most up-regulated mRNA was forkhead box protein I3 (FOXI3, FC 130.34), whereas the most down-regulated mRNA was hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 (HSD3B1, FC −210.81).
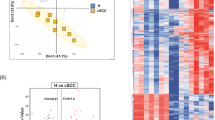
Scatterplot analysis showed variations in the lncRNA and mRNA expression profiles in the BCC samples compared with the control samples (Fig. 1a, b). To visualize the relationship between the fold changes and the statistical significance of differentially expressed lncRNAs and mRNAs, volcano plots were constructed (Fig. 2a, b). Furthermore, the lncRNA and mRNA expression patterns across samples were distinguishable in the heat map generated by hierarchical clustering (Fig. 3a, b). Correlation analysis of lncRNA and mRNA expression showed high correlations within the two groups of BCC and control samples (Pearson’s correlation coefficient r > 0.9). The results from the correlation analyses are summarized in Fig. 4a, b.

Scatterplots of lncRNA (a) and mRNA (b) showing normalized signal values (log2 scaled) for BCC versus control. All points above and below the green fold change lines indicate a greater-than-2.0-fold change of lncRNA/mRNA between BCC versus control

Volcano plots showing differentially expressed lncRNAs (a) and mRNAs (b); red squares represent differentially expressed lncRNAs. The two vertical lines represent the filtering criteria of FC ≥ 2

Cluster analysis of lncRNA (a) and mRNA (b) expression shows a strong similarity within the two groups (BCC and non-lesional skin) with clustering. The color scale reflects the signal intensity converted to log2 and ranges from blue (low intensity) to white (moderate intensity) to red (strong intensity). The dendrogram (left) reflects the hierarchical similarity

Heat map of correlation coefficient r analysis (BCC vs. control) for lncRNA (a) and mRNA expression (b). The color scale on the left reflects the correlation of samples and runs from white (low correlation) to blue (medium correlation) to red (high correlation) (BCC basal cell carcinoma)
GO and KEGG pathway analysis
To determine the potential role of differentially expressed mRNAs in BCC, GO categories of cc, bp, and mf were analyzed as previously described [35, 36]. Enriched GO terms “extracellular matrix” (cc), “anatomical structure morphogenesis” (bp), and “metal ion binding” (mf) were the most significantly up-regulated mRNAs (p ≤ 0.05; FDR ≤ 0.05) (Fig. 5). The analysis of the down-regulated mRNAs showed that the “membrane region” (cc), “lipid metabolic process” (bp), and “oxidoreductase activity” (mf) were the most significant (p ≤ 0.05; FDR ≤ 0.05). The KEGG pathway analysis showed that “Focal adhesion” “Extracellular matrix (ECM) receptor interaction,” and the cell cycle-regulating “PI3K-Akt signaling pathway” showed the highest enrichment scores in the group of up-regulated mRNAs; specifically, “Peroxisome,” “Fatty acid metabolism,” and “Biosynthesis of unsaturated fatty metabolism” showed the highest enrichment scores in the groups of down-regulated mRNAs (p ≤ 0.05; FDR ≤ 0.05).

Differentially expressed mRNAs functionally classified by GO analysis (5A-5C; filtering criteria p ≤ 0.05 with a FC ≥ 2.0) and KEGG pathway analysis (5D). Up- or down-regulated mRNAs in BCC were analyzed in three GO categories: a biological process (bp), b cellular component (cc), and c molecular function (mf)
Discussion
The role of lncRNAs in cancer has recently been addressed in numerous studies in a variety of malignant tissues. In addition, differential expression functional studies have shown the enormous potential of lncRNA-based therapeutic mechanisms. Xing et al. showed that lncRNA participates in the direct coordination of protein recruitment and the indirect regulation of transcription factors, which can be targeted by lncRNA silencing [37]. In a mouse model, they showed that knockdowns of BCAR4 lncRNA significantly reduced breast cancer metastasis. Although BCC is the most frequently occurring tumor in humans being semi-malignant, slow growing, and locally infiltrating, there has been no global investigation of differential lncRNA expression in BCC [38, 39]. The present study showed that a variety of lncRNAs were differentially expressed in BCC and, similar to miRNAs, may play an important role in BCC pathology. The gene AC073135.3 encoding the two most up-regulated lncRNAs, ENST00000411596 and ENST00000453982, has been found to be up-regulated in lung adenocarcinoma and glioma cells [40]. The gene RP6-24A23.7 splicing lncRNA sequence ENST00000564206 was predicted to be associated with lung neoplasm, glioma, colorectal neoplasm, melanoma, pancreatic neoplasm, neuroblastoma, and breast neoplasm based on a model of LncRNA Functional Similarity Calculations based on the information of MiRNA (LFSCM), which calculates functional similarities of lncRNA and disease-specific lncRNA-miRNA interactions [41]. Interestingly, lncRNAs, similar to circular RNAs, influence gene expression by acting as miRNA sponges [10]. They can contain multiple miRNA-binding sites capable of binding target miRNAs by complimentary base pairing, which regulates mRNA expression.
ENST00000442037 is spliced from the gene H19, which has been shown to play an important role in gastric cancer through its mature product hsa-miR-675, which targets the tumor suppressor Runt domain transcription factor1 (RUNX1) [42, 43]. For future studies, it would be interesting to determine if a similar lncRNA-miRNA-tumor suppressor gene axis can be identified for BCC. ENST00000500092 spliced from RP11-519 M16.1 was predicted to target hsa-miR-19b-1, a member of oncogenic oncomiR-1 (miRNa-17-92), and hsa-miR-106a, which shares seed region identity with hsa-miR-106b [44, 45]. In a previous study, we showed that hsa-miR-19b-1 and hsa-miR-106b are both significantly up-regulated in BCC [4].
ENST00000522183 spliced from RP11-697 M17 uses hsa-miR-130a and hsa-miR-145-3p as target sequences. hsa-miR-130a was up-regulated, and hsa-miR-145-3p down-regulated, in a previous BCC miRNA expression profiling study [4]. AC073135.3 was up-regulated in lung adenocarcinoma [40]. LINC00340 (cancer susceptibility candidate 15, CASC15) is associated with neuroblastoma and is increased during melanoma progression [46]. It has further been shown to be an independent predictor of disease recurrence in melanoma patients with stage III lymph node metastasis and plays a key role in switching between proliferative and invasive states in melanoma cells [47]. KC6 splicing NR_002838 was described in keratoconus, which is a degenerative proliferative disorder of the eye characterized by conical-shaped cornea epithelium [48].
Hombach et al. reviewed lncRNAs involved in epidermal homeostasis and disease [10]. When comparing previously described relevant skin-associated lncRNAs with our data set, several important similarities were found. The SPRY4 intronic transcript 1 (SPRY4-IT1) was up-regulated in our study and is up-regulated in several different tumors, including melanoma, gastric cancer, and breast and prostate cancer [10]. Antisense noncoding RNA in the INK4 locus (ANRIL), which is also known as CDKN2B antisense RNA 1, is associated with a variety of malignancies, such as melanoma, breast tumors, small cell lung cancer, hepatocellular carcinoma, and cervical cancer [49, 50]. In our data set, CDKN2B was significantly up-regulated; however, ANRIL was not differentially expressed (p < 0.05). Tissue differentiation-inducing nonprotein-coding RNA (TINCR or PLAC2) was up-regulated in differentiating keratinocytes, together with Staufen1 building a protein-lncRNA complex that regulates genes required for terminal differentiation and maintaining high levels of pivotal epidermal differentiation genes, such as fillagrin and loricrin [51–53]. In our study, it was up-regulated in BCC; however, it did not reach statistical significance (p > 0.05). Differentiation antagonizing nonprotein coding RNA (DANCR or ANCR) suppressed epidermal differentiation in progenitors and was slightly down-regulated in BCC (p > 0.05) [14]. Recently, both TINCR and ANCR were shown to regulate the v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF, also known as c-maf) and the v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB), which were shown to bind and control the expression of known epidermal differentiation transcription factor genes, such as GRHL3, ZNF750, KLF4, and PRDM1 [15]. The lncRNAs TINCR and ANCR regulate MAF/MAFB-induced epidermal transcription factors representing an lncRNA transcription factor network that is essential for epidermal differentiation and is possibly involved in epithelial skin cancer, such as BCC.
The lncRNA psoriasis associated nonprotein coding RNA induced by stress (PRINS) regulates interferon-induced protein 6-16 (G1P3), exhibits anti-apoptopic effects in keratinocytes, and is down-regulated in psoriasis [54]. PRINS was down-regulated in BCC, but this change did not reach statistical significance (p > 0.05). The HOXA distal transcript antisense RNA (HOTTIP) important for transcriptional activation can activate HoxA genes by recruiting histone-modifying enzymes in an intracellular phenomenon described as locus control [55]. In vitamin D receptor (VDR) deleted mouse keratinocytes, which are more prone to UV-induced skin cancer development, the oncogenic lncRNA HOTTIP was significantly increased [56]. HOTTIP was also increased in BCC, but this increase did not reach statistical significance (p > 0.05).
Conclusions
In conclusion, the results of this study showed the first evidence for lncRNA and mRNA differential expression in BCC compared to NLES (control).
Differential expression of lncRNAs in BCC was noticeable and therefore suggests a potentially important role for lncRNAs in BCC formation. The differentially expressed lncRNAs described in this work represent a foundation for future functional studies which could potentially lead to lncRNA-based therapy of advanced or metastatic BCC.
References
Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001;2:986–91.
Sand M. The pathway of miRNA maturation. Methods Mol Biol. 2014;1095:3–10.
Sand M, Gambichler T, Sand D, Skrygan M, Altmeyer P, Bechara FG. MicroRNAs and the skin: tiny players in the body’s largest organ. J Dermatol Sci. 2009;53:169–75.
Sand M, Skrygan M, Sand D, Georgas D, Hahn SA, Gambichler T, et al. Expression of microRNAs in basal cell carcinoma. Br J Dermatol. 2012;167:847–55.
Sand M, Gambichler T, Skrygan M, Sand D, Scola N, Altmeyer P, et al. Expression levels of the microRNA processing enzymes Drosha and dicer in epithelial skin cancer. Cancer Investig. 2010;28:649–53.
Sand M, Skrygan M, Georgas D, Arenz C, Gambichler T, Sand D, et al. Expression levels of the microRNA maturing microprocessor complex component DGCR8 and the RNA-induced silencing complex (RISC) components argonaute-1, argonaute-2, PACT, TARBP1, and TARBP2 in epithelial skin cancer. Mol Carcinog. 2012;51:916–22.
Sand M, Sand D, Altmeyer P, Bechara FG. MicroRNA in non-melanoma skin cancer. Cancer Biomarkers : Sect Dis Markers. 2012;11:253–7.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Guo X, Gao L, Wang Y, Chiu DK, Wang T, Deng Y et al. Advances in long noncoding rnas: Identification, structure prediction and function annotation. Brief Funct Genomics 2015, in press
Hombach S, Kretz M. The non-coding skin: exploring the roles of long non-coding RNAs in epidermal homeostasis and disease. Bioessays. 2013;35:1093–100.
Ezkurdia I, Juan D, Rodriguez JM, Frankish A, Diekhans M, Harrow J, et al. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum Mol Genet. 2014;23:5866–78.
Li X, Wu Z, Fu X, Han W. LncRNAs: insights into their function and mechanics in underlying disorders. Mutat Res Rev Mutat Res. 2014;762:1–21.
Isin M, Dalay N. LncRNAs and neoplasia. Clin Chim Acta. 2015;444:280–8.
Kretz M, Webster DE, Flockhart RJ, Lee CS, Zehnder A, Lopez-Pajares V, et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev. 2012;26:338–43.
Lopez-Pajares V, Qu K, Zhang J, Webster DE, Barajas BC, Siprashvili Z, et al. A LncRNA-MAF:MAFB transcription factor network regulates epidermal differentiation. Dev Cell. 2015;32:693–706.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58.
Mazar J, Sinha S, Dinger ME, Mattick JS, Perera RJ. Protein-coding and non-coding gene expression analysis in differentiating human keratinocytes using a three-dimensional epidermal equivalent. Mol Genet Genomics : MGG. 2010;284:1–9.
Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: a potential novel class of cancer biomarkers. Front Genet. 2015;6:145.
Hajjari M, Khoshnevisan A, Shin YK. Molecular function and regulation of long non-coding RNAs: paradigms with potential roles in cancer. Tumour Biol. 2014;35:10645–63.
Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–46.
Harrow J, Denoeud F, Frankish A, Reymond A, Chen CK, Chrast J, et al. GENCODE: producing a reference annotation for encode. Genome Biol. 2006;7(1):1–9. S4.
Pang KC, Stephen S, Dinger ME, Engstrom PG, Lenhard B, Mattick JS. RNAdb 2.0-—an expanded database of mammalian non-coding RNAs. Nucleic Acids Res. 2007;35:D178–82.
Dinger ME, Pang KC, Mercer TR, Crowe ML, Grimmond SM, Mattick JS. NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 2009;37:D122–6.
Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. LncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39:D146–51.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncodingRNA reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27.
Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–5.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23.
Luo X, Shi Q, Gu Y, Pan J, Hua M, Liu M, et al. LncRNA pathway involved in premature preterm rupture of membrane (PPROM): an epigenomic approach to study the pathogenesis of reproductive disorders. PLoS One. 2013;8, e79897.
Gene Ontology C. Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56.
Consortium TGO. Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84.
Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–27.
Sokal RR, Michener CD. A statistical method for evaluating systematic relationships. Univ Kansas Sci Bull. 1958;28:1409–38.
Gene Ontology C. The gene ontology project in 2008. Nucleic Acids Res. 2008;36:D440–4.
Gene Ontology C. The gene ontology in 2010: extensions and refinements. Nucleic Acids Res. 2010;38:D331–5.
Xing Z, Lin A, Li C, Liang K, Wang S, Liu Y, et al. LncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell. 2014;159:1110–25.
Miller SJ. Biology of basal cell carcinoma (part I). J Am Acad Dermatol. 1991;24:1–13.
Miller SJ. Biology of basal cell carcinoma (part II). J Am Acad Dermatol. 1991;24:161–75.
Wang Y, Chen W, Chen J, Pan Q, Pan J. LncRNA expression profiles of EGFR exon 19 deletions in lung adenocarcinoma ascertained by using microarray analysis. Med Oncol. 2014;31:137.
Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015;5:13186.
Zhuang M, Gao W, Xu J, Wang P, Shu Y. The long non-coding RNA h19-derived miR-675 modulates human gastric cancer cell proliferation by targeting tumor suppressor RUNX1. Biochem Biophys Res Commun. 2014;448:315–22.
Li H, Yu B, Li J, Su L, Yan M, Zhu Z, et al. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014;5:2318–29.
Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–32.
Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1a and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–74.
Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M, et al. CASC15-s is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. 2015;75:3155–66.
Lessard L, Liu M, Marzese DM, Wang H, Chong K, Kawas N et al. The casc15 long intergenic noncoding rna locus is involved in melanoma progression and phenotype switching. J Invest Dermatol 2015;135(10):2464–74.
Rabinowitz YS, Dong L, Wistow G. Gene expression profile studies of human keratoconus cornea for NEIBank: a novel cornea-expressed gene and the absence of transcripts for aquaporin 5. Invest Ophthalmol Vis Sci. 2005;46:1239–46.
Naemura M, Murasaki C, Inoue Y, Okamoto H, Kotake Y. Long noncoding RNA ANRIL regulates proliferation of non-small cell lung cancer and cervical cancer cells. Anticancer Res. 2015;35:5377–82.
Iranpour M, Soudyab M, Geranpayeh L, Mirfakhraie R, Azargashb E, Movafagh A et al. Expression analysis of four long noncoding rnas in breast cancer. Tumour Biol 2015, in press
Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–5.
Wan DC, Wang KC. Long noncoding RNA: significance and potential in skin biology. Cold Spring Harb Perspect Med. 2014;4.
Kretz M. TINCR, staufen1, and cellular differentiation. RNA Biol. 2013;10:1597–601.
Szegedi K, Sonkoly E, Nagy N, Nemeth IB, Bata-Csorgo Z, Kemeny L, et al. The anti-apoptotic protein G1P3 is overexpressed in psoriasis and regulated by the non-coding RNA, PRINS. Exp Dermatol. 2010;19:269–78.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4.
Jiang YJ, Bikle DD. LncRNA profiling reveals new mechanism for VDR protection against skin cancer formation. J Steroid Biochem Mol Biol. 2014;144(Pt A):87–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Financial disclosure
All authors hereby disclose any commercial associations that may pose or create a conflict of interest with the information presented in this manuscript. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper. Daniel Sand was supported by the Heed Ophthalmic Foundation.
Ethics
This study conformed to local requirements following ethical and investigational committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of human subjects participating in medical research (Ethical Review Board of Ruhr-University Bochum, Germany).
Rights and permissions
About this article

Cite this article
Sand, M., Bechara, F.G., Sand, D. et al. Long-noncoding RNAs in basal cell carcinoma. Tumor Biol. 37, 10595–10608 (2016). https://doi.org/10.1007/s13277-016-4927-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-4927-z