
Abstract
Neuroglioma is a complex neuroglial tumor involving dysregulation of many biological pathways at multiple levels. Here, we aim to screen differentially expressed miRNAs (DEMs) as well as the functions and pathways of their target genes in neuroglioma. miRNA high-throughput sequencing data were downloaded from The Cancer Genome Atlas (TCGA), and then the DEMs were subjected to perform principal component analysis (PCA) based on their expression values. Following that, Targetscan software was used to predict the target genes, and enrichment analysis and pathway annotation of these target genes were done by DAVID and KEGG, respectively. Finally, survival analysis between the DEMs and patients’ survival time was done, and the miRNAs with prediction potential were obtained. A total of 33 DEMs were obtained, among which 25 miRNAs were upregulated including hsa-mir-675, hsa-mir-196a-1, and hsa-mir-196a-2, while eight miRNAs were downregulated including hsa-mir-1911, hsa-mir-1264, and hsa-mir-1298. Five miRNAs with diagnostic and preventive potentials were significantly correlated with survival time, including has-mir-155, has-mir-199b, has-mi-10a, has-mir-1274b, and has-mir-455. The target genes of miRNA identified in this study played important roles in tumor signaling pathways, and their detailed functions could be further studied so as to explore novel neuroglioma therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroglioma is one of the most common primary brain tumors in adults, which affects more males than females; it is also one of the main causes of cancer-related death worldwide. Although great progresses in diagnosis and management of neuroglioma have been made in recent years, its exact molecular mechanisms remain poorly understood, which limits the prognosis and therapy. With the aim to study the detailed molecular mechanism of neuroglioma and develop effective therapies, it is of great importance to explore and identify new biological markers.
MicroRNAs (miRNAs) are a class of small non-coding (18–25 nucleotides), naturally occurred endogenous, noncoding RNA molecules that regulate the translation of messenger RNAs (mRNAs) by binding to the target mRNAs [1]. miRNAs are thought to play critical regulatory roles in many physiological processes such as cell proliferation, differentiation, apoptosis, and cell death [1, 2]. Furthermore, the role of miRNAs has been well elaborated in various human cancers, and abnormal miRNA expression can disturb various cell signaling pathways during cancer development. The expression changes of miRNAs have been observed in a variety of human tumors such as lung cancer [3], breast cancer [4], or glioblastoma [5]. The expression differences can not cause carcinogenesis directly; however, these changes may regulate several key genes during the process of tumor pathogenesis and play considerable roles in the prognosis and therapy of tumors. Take microRNA-21 as an example, which has been reported overexpressed in neuroglioma. microRNA-21 can target PDCD4 at the posttranscriptional level and regulates cell proliferation and invasion, indicating that it may serve as a novel therapeutic target in neuroglioma [6]. Previous studies have proved that miRNAs played critical roles in the initiation, development, invasion, and metastasis of tumors, yet their underlying molecular mechanisms still remain poorly understand. It is of great importance to identify new and specific molecular markers that may contribute to the early diagnosis or prognosis of neuroglioma.
This study screened differentially expressed miRNAs (DEMs) in neuroglioma and analyzed the target genes of these DEMs by DAVID. Through performing survival analysis between the DEMs and patients’ survival time, we hope to find miRNAs with prediction potential and explore molecular markers for early diagnosis and treatment of neuroglioma.
Materials and methods
miRNA microarray data and patient information
The miRNA expression data and the corresponding patients’ medical information were obtained from The Cancer Genome Atlas (TCGA) database, including a total of 475 samples (381 neuroglioma samples and 94 normal samples). The miRNA expression data were sequenced by Illumina HiSeq system, while the standardized miRNA data were level three.
The miRNA data were removed when expression values were zero. The level three miRNA data consist of a total of 1046 comments for miRNA expression values. The level three miRNA data had already been standardized between samples using generalized linear model in R language Limma package to eliminate the batch effects between samples.
DEMs screening
The DEMs between normal and neuroglioma tissue samples were screened by SAMR [7] package in R software. LogFC and p represented the differential expression degree, while Log2FC indicated the differential expression degree of miRNA between differentially expressed tumor samples and normal samples. Downregulated and upregulated miRNAs were expressed as logFC <−1 and logFC >1, respectively, both with FDR <0.05. Principal component analysis method was conducted to distinguish the DEMs between neuroglioma and normal tissue samples efficiently.
Survival analysis
All medical information for patients was summarized and subjected to perform statistical analysis in order to determine the cutoff value of medical information. The distribution of survival time and the differences of survival ability under various diseased states were studied and defined by Kaplan-Meier and log-rank respectively. Moreover, the relationship between DEMs and survival time of patients were explored using Univariate Cox regression model. The original data were arranged sequentially as survival days, survival state (status, death was 1 while survival was 2), cumulative survival, standard error of the survival rate, cumulative event, and number remaining of each sample. Eventually, the survival rates of patients were obtained.
Screening and conformation of the molecular markers for miRNAs
The expression profiling of miRNAs in training group was analyzed by Cox regression model with the aim to acquire survival-related miRNAs (P < 0.01) and five miRNAs significantly associated with survival (P < 0.005). Patients’ survival abilities could be predicted by a multivariable Cox regression model constructed based on the five miRNAs. As each miRNA parameter possessed a Cox regression correlation coefficient, thus, every patient had a risk factor. The patients with higher risk scores exhibited weaker viability comparing to those with lower risk scores in the light of this model. The median risk score may be considered, as the boundary was calculated. The risk score above the boundary was defined as high risk, while it was low risk below the boundary. The survival time distribution for each variable was observed by Kaplan-Meier model, and the significances among various classifications under the same variable were determined utilizing log-rank detection method.
Analysis of the target genes of five miRNAs
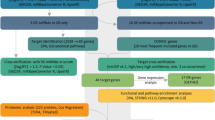
The target genes of the DEMs in Mirtarbase [8] database were extracted at first, and then GO functional annotation and KEGG pathways of these target genes were performed by DAVID software [9]. According to each GO value, a P value of function enrichment and a corrected P value after multiple testing (Benjamini correction) would be calculated by DAVID (the detailed flowcharts are shown in Fig. 1).

Flowcharts for analyzing differential miRNAs and their target genes
Results
Expression differences of miRNAs among various samples
The differences between normal and cancer samples were observed by principal component analysis and cluster analysis. The normal samples and tumor samples are separated in Fig. 2. However, there was a normal sample distributed in the upper right, indicating that an error may occur, so this sample needed to be removed in the following process.

Principal component analysis of the miRNA expression values. Principal component analysis was complicated according to the miRNA expression differences between normal and cancer samples. The horizontal axis represented that the first principal component accounted for 19 %, while the vertical axis represented that the second principal component accounted for 15 %. The red dots and black spots indicated cancer samples and normal samples, respectively. From the figure, one normal sample was distributed on the top right, and we have removed the outlier from the following analysis. We could clearly observe that normal samples gathered on the top left, while cancer samples gathered on the bottom right, indicating that there were significant differences between the two samples
Differential expression analysis of miRNA
We obtained a total of 33 DEMs between normal and neuroglioma samples, including 25 upregulated and 8 downregulated miRNAs. The upregulated miRNAs including hsa-mir-675, hsa-mir-196a-1, and hsa-mir-196a-2 accounted for 75.8 % of the total differential miRNAs, while the downregulated miRNAs including hsa-mir-1911, hsa-mir-1264, and hsa-mir-1298 accounted for 24.2 % (Table 1).
Survival analysis
From the medical information of patients in Table 2, we found that the censoring rate 75.33 % was relatively high, indicating that there were still many patients alive at the end of the study or they directly went away. A survival curve was drawn according to the patients’ survival time (Fig. 3). From Fig. 3, we understood that although there was a high cutoff value, the medical information Table 2 was enough and suitable to predict the biological targets of miRNAs in the following study.

The overall survival status changes of patients. The survival curves were drawn according to the patients’ survival time. The horizontal axis showed survival time, and the vertical axis showed survival rates; the red crosses on curve were cutoff values. See from the figure, patients’ viability decreased gradually over time
Kaplan and log-rank methods were taken to verify the relationship between the survival time and various variables (including age, sex, T stage, R stage, M stage, N stage, and tumor stage), and the results are demonstrated in Fig. 4. Significant differences were found between different tumor status and the overall survival time. Figure 4 demonstrates that univariate cox regression model could be applied to analyze the differences between miRNA and survival time of patients under each different state. The correlation coefficients are demonstrated in heat map of Fig. 5. The molecular markers of miRNAs were selected with P < 0.1, and there were significant results at least in two separate categories.

The variations of patients’ survival status in different classifications. a–f represented the relationships between age, gender, T stage, R stage, M stage, N stage, tumor stage, and survival time, respectively. P value of Log rank test was shown in the lower left corner of each figure

Predictive capabilities of miRNAs under different tumor status. The abscissa and the ordinate showed different tumor status and different miRNAs respectively. This heat map showed the capacity that the 140 differentially expressed miRNAs of tumor affected the survival under various status. Red represented strong positive correlations between miRNA expression and survival time, while green represented strong negative correlations
The miRNAs associated with patients’ overall survival in different tumor status were filtered through univariate survival analysis (Fig. 5). Five miRNAs including has-mir-155, has-mir-199b, has-mir-10a, hsa-mir-1274b, and has-mir-455 that may be considered as biomarkers were selected.
Construction of predictive disease model
Cox multivariate regression model was applied to construct a mathematical model for miRNA and survival time using data in training group, the prognosis formula was as follows: prognostic score = (1.15 × expression level of hsa-mir-155) + (0.58 × expression level of hsa-mir-199b) + (0.2 × expression level of hsa-mir-10a) + (−1.97 × expression level of hsa-mir-1274b) + (0.3 × expression level of hsa-mir-455). Among which, four miRNAs belonged to risky type, and one belonged to protective type.
The differences between patients in high-risk and low-risk groups were analyzed by Log-rank test, and results are shown in Fig. 6. There were significant differences between high-risk and low-risk patients in training group, also the same with test group, indicating that this model was feasible in actual prediction process. The prediction results of miRNA in patients are displayed in Fig. 7, which showed the expression differences of miRNA between high-risk and low-risk patients. Moreover, we could know from the results that the lower the risk coefficient, the longer the survival time.

Determination of patients’ survival risk by Kaplan-Meier based on expression levels of five miRNAs. The abscissa showed the survival time, while the ordinate showed the survival rate. Red and blue represented high-risk and low-risk patients, respectively. P value at the lower left corner indicated the differences between two risk groups

Analysis of the risk coefficients predicted by the five miRNAs. a Risk coefficients analysis of training set data, b heat map for expression levels of five miRNAs in training set, c, d the relationship between the patients’ survival time and risk coefficients, and e, f distribution of risk coefficients
Analysis of the target genes for five miRNAs
A total of 4479 target genes were predicted by prediction software Targetscan. Then, these target genes were subjected to conduct GO annotation and KEGG pathway analysis by David. GO annotation results demonstrated that the main functions of these target genes were associated with protein amino acid phosphorylation and positive regulation of gene expression, while the results of KEGG analysis showed that they were mainly related to endocytosis and pathways in cancer (Fig. 8).

GO and KEGG analysis of the target genes of differentially expressed miRNAs. a GO analysis of the target gene, and b KEGG analysis of the target gene. The abscissa represented significant degree, while the vertical axis represented functional annotation; the greater the significant degree, the greater correlation between the target gene and annotation
Discussion
In this study, we screened a total of 33 differentially expressed miRNAs between normal and neuroglioma samples. Then, according to the medical information of patients, we predicted the miRNAs that could be considered as molecular targets, and five miRNAs including has-mir-155, has-mir-199b, has-mir-10a, has-mir-1274b, and has-mir-455 were finally selected.
A previous study has reported that some neuronal miRNA may show context-dependent roles in neurological diseases through regulating the spatial and temporal expression levels of protein-coding genes, such as miR199b [10]. miR199b can regulate cell proliferation, survival, and death by targeting hypoxia-inducible factor 1 alpha (HIF-1α) [11, 12]. HIF-1α, a vital transcription factor which regulates gene expression mainly in the brain to response to the decreases in oxygen availability [13, 14], plays an important role in cell regeneration and survival. In addition, HIF-1α is found to be involved in some pathophysiologic processes during disease development, including neurodegenerative diseases, and tumorigenesis [15, 16]. Several recent studies have proved that HIF-1α is a target of miR199b, indicating that miR199b may represent a novel prognostic or therapeutic marker for neuroglioma. Also, it is reported that overexpression of miR-155 can accelerate the growth of tumor cell lines in vivo [17]. The upregulation of mir-155 in several types of cancers like B-cell lymphoma combining with its functional studies suggests that it is oncogenic [18, 19]. Furthermore, a recent study shows that miR-155 has mutator activity [20] and several known target genes of miR-155 correlate with apoptotic and proliferative response and contribute to tumor development [21, 22].
In order to further analyze the selected miRNAs and explore the possible pathogenesis of neuroglioma, the target genes of these miRNAs were also predicted. A total of 4479 target genes were obtained. The main functions of these target genes were associated with protein amino acid phosphorylation and positive regulation of gene expression, while the major pathways of these genes were enriched in endocytosis and pathways in cancer through GO annotation and KEGG analysis. As we all know, the reversible protein phosphorylation regulates nearly every aspect of cell life, and phosphorylation or dephosphorylation catalyzed by protein kinases and protein phosphatases, respectively, can modify the function of a protein in almost every conceivable way [23]. It is reported that nearly 30 % of the proteins encoded by the human genome have covalently bound phosphate; moreover, abnormal phosphorylation is considered as a cause for various human diseases. Numerous naturally occurred toxins and tumor promoters exert their effects through targeting particular protein kinases and phosphatases [24]. Therefore, we speculated that these target genes may be also considered as useful targets for neuroglioma. As for the pathways these genes enriched in, we take endocytosis as an example. Endocytosis is regarded as a signaling pathway, triggered by the activation of surface receptors and leading to their internalization, and targets them for degradation in the lysosome [25]. Recently, there are studies indicating that endocytosis may play a role in cancer [26]. Some connections between endocytosis and cancer include the following: (1) As endocytosis is an attenuator of signaling, therefore, it is a potential candidate as a tumor suppressor pathway; (2) endocytosis is involved in several pathways leading to the activation of certain receptors of established relevance to cancer, such as Notch. In other words, these target genes may be involved in the pathogenesis of neuroglioma and could be considered as biomarkers for the treatment of neuroglioma.
In summary, we screened differentially expressed miRNAs between normal and neuroglioma samples in this study and selected five miRNAs including has-mir-155, has-mir-199b, has-mir-10a, has-mir-1274b, and has-mir-455 with potential of predictive ability in neuroglioma. In addition, we analyzed the target genes of these five miRNAs, which may provide new insights into cancer development and find new potential biomarkers and therapeutic targets. Although further studies are still needed, the results in this study could help understand the pathogenesis of neuroglioma.
References
Schickel R, Boyerinas B, Park S, Peter M. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–74.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Yu S-L, Chen H-Y, Chang G-C, Chen C-Y, Chen H-W, Singh S, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48–57.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33.
Zhang J-G, Wang J-J, Zhao F, Liu Q, Jiang K, Yang G-H. MicroRNA-21 (mir-21) represses tumor suppressor pten and promotes growth and invasion in non-small cell lung cancer (nsclc). Clin Chim Acta. 2010;411:846–52.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci. 2001;98:5116–21.
Hsu S-D, Lin F-M, Wu W-Y, Liang C, Huang W-C, Chan W-L, et al. Mirtarbase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2011;39:D163–9.
Dennis Jr G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. David: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3.
Jiang G, Zhou R, He X, Shi Z, Huang M, Yu J, et al. Expression levels of microRNA-199 and hypoxia-inducible factor-1 alpha in brain tissue of patients with intractable epilepsy. Int J Neurosci. 2014;1–29.
Wang C, Song B, Song W, Liu J, Sun A, Wu D, et al. Underexpressed microRNA‐199b‐5p targets hypoxia‐inducible factor‐1α in hepatocellular carcinoma and predicts prognosis of hepatocellular carcinoma patients. J Gastroenterol Hepatol. 2011;26:1630–7.
Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of mir-199a derepresses hypoxia-inducible factor-1α and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–86.
Aro E, Khatri R, Gerard-O'Riley R, Mangiavini L, Myllyharju J, Schipani E. Hypoxia-inducible factor-1 (hif-1) but not hif-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J Biol Chem. 2012;287:37134–44.
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors hif-1α and hif-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–21.
Alidoosti M, Ghaedi M, Soleimani A, Bakhtiyari S, Rezvanfard M, Golkhu S, et al. Study on the role of environmental parameters and hif-1a gene polymorphism in coronary collateral formation among patients with ischemic heart disease. Clin Biochem. 2011;44:1421–4.
Cao WD, Kawai N, Miyake K, Zhang X, Fei Z, Tamiya T. Relationship of 14-3-3zeta (zeta), hif-1alpha, and vegf expression in human brain gliomas. Brain Tumor Pathol. 2014;31:1–10.
Chang S, Wang R-H, Akagi K, Kim K-A, Martin BK, Cavallone L, et al. Tumor suppressor brca1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–82.
O'Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–94.
Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, et al. Bic and mir‐155 are highly expressed in hodgkin, primary mediastinal and diffuse large b cell lymphomas. J Pathol. 2005;207:243–9.
Yamamoto M, Kondo E, Takeuchi M, Harashima A, Otani T, Tsuji-Takayama K, et al. Mir-155, a modulator of foxo3a protein expression, is underexpressed and cannot be upregulated by stimulation of hozot, a line of multifunctional treg. PLoS One. 2011;6:e16841.
Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, et al. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting foxo3a in breast cancer. J Biol Chem. 2010;285:17869–79.
Jiang S, Zhang H-W, Lu M-H, He X-H, Li Y, Gu H, et al. MicroRNA-155 functions as an oncomir in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–27.
Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–30.
MacKintosh C, Beattie KA, Klumpp S, Cohen P, Codd GA. Cyanobacterial microcystin-lr is a potent and specific inhibitor of protein phosphatases 1 and 2a from both mammals and higher plants. FEBS Lett. 1990;264:187–92.
Mellman I, Yarden Y. Endocytosis and cancer. Cold Spring Harbor Perspectives Biol. 2013;5:a016949.
Lanzetti L, Di Fiore PP. Endocytosis and cancer: an ‘insider’ network with dangerous liaisons. Traffic. 2008;9:2011–21.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, H., Wang, Y. Five miRNAs considered as molecular targets for predicting neuroglioma. Tumor Biol. 37, 1051–1059 (2016). https://doi.org/10.1007/s13277-015-3898-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3898-9