
Abstract
In the current study, we tested the potential anti-pancreatic cancer activity of a novel hydroxamate-based histone deacetylase (HDAC) inhibitor ST-3595. We showed that ST-3595 exerted potent anti-proliferative and cytotoxic activities against both established pancreatic cancer cell lines (PANC-1, AsPC-1, and Mia-PaCa-2), and patient-derived primary cancer cells. It was, however, generally safe to non-cancerous pancreatic epithelial HPDE6c7 cells. ST-3595-induced cytotoxicity to pancreatic cancer cells was associated with significant apoptosis activation. Reversely, the pan caspase inhibitor z-VAD-fmk and the caspase-8 inhibitor z-ITED-fmk alleviated ST-3595-mediated anti-pancreatic cancer activity in vitro. For the mechanism study, ST-3595 inhibited HDAC activity, and induced mitochondrial permeability transition pore (MPTP) opening in pancreatic cancer cells. Inhibition of MPTP, by cyclosporin A, sanglifehrin A, or by cyclophilin-D (Cyp-D) siRNA knockdown, dramatically inhibited ST-3595-induced pancreatic cancer cell apoptosis. Meanwhile, we found that a low concentration of ST-3595 dramatically sensitized gemcitabine-induced anti-pancreatic cancer cell activity in vitro. In vivo, ST-3595 oral administration inhibited PANC-1 xenograft growth in nude mice, and this activity was further enhanced when in combination with gemcitabine. In summary, the results of this study suggest that targeting HDACs by ST-3595 might represent as a novel and promising anti-pancreatic cancer strategy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is an extremely aggressive malignancy with poor prognosis [1–3]. It is characterized by rapid disease progression without specific clinical symptoms, and its 5-year overall survival is often less than 5 % [1–3]. Gemcitabine is the only approved single-agent chemotherapeutic drug for pancreatic cancer [4]. However, pancreatic cancer is often resistant to gemcitabine and other known anti-cancer drugs [1–3]. Groups around the world are working extremely hard on exploring alternative and more efficient anti-pancreatic cancer agents and gemcitabine sensitizers [3, 5, 6].
Histone deacetylases (HDACs), often deregulated in pancreatic cancer and other solid tumors [7, 8], are implicated in regulating of molecules in growth regulatory and/or apoptotic pathways [7, 8]. Activation of HDAC pathways in pancreatic cancer cells [9, 10] will likely lead to the failure to undergo apoptosis, causing cancer resistance [7, 8]. Thus, HDAC inhibitors (HDACis) have been developed [11]. At least six structurally distinct HDACis have been developed thus far, including short-chain fatty acids, hydroxamic acids, cyclic peptides, benzamides, electrophilic ketones, and hybrid molecules [11].
Many of these HDACis have displayed significant anti-cancer activities, either alone or in combination with other therapeutic agents [10, 11]. These HDACis also displayed promising results in pancreatic cancer models [9, 10]. In the current study, we investigated the potential activity of ST-3595, a novel hydroxamate-based HDAC inhibitor, against pancreatic cancer cells in vitro and in vivo. The potential gemcitabine-sensitization effect of ST-3595 was also evaluated.
Material and methods
Chemicals and reagents
Gemcitabine, vorinostat (SAHA), sanglifehrin A (SfA), and cyclosporine A (CsA) were obtained from Sigma (Sigma, St. Louis, MO); Apoptosis inhibitors z-VAD-fmk and z-ITED-fmk were obtained from Calbiochem (Shanghai, China). Anti-tubulin, cyclophilin-D (Cyp-D), and HDAC-1/-2/-4/-6 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ST-3595 was obtained from Sigma-Tau (Pomezia, Italia).
Cell culture
PANC-1, AsPC-1, and Mia-PaCa-2 pancreatic cancer cell lines were purchased from the Shanghai Institute of Biological Science, Chinese Academy of Science (Shanghai, China). Cancer cells were cultured in RPMI/DMEM medium (Invitrogen, Shanghai, China), supplemented with 10 % fetal bovine serum (FBS, Invitrogen) with antibiotics in a CO2 incubator at 37 °C. The non-transformed pancreatic epithelial cell line HPDE6c7 (non-cancerous line) was purchased from Shanghai Rui-Lu Biotech (Shanghai, China) [12]. HPDE6c7 cells were cultivated in DMEM supplemented with 10 % FBS and necessary antibiotics.
Patient-derived pancreatic cancer cell culture
Surgery-isolated pancreatic cancer tissues from informed-consent patients were thoroughly washed in DMEM and 1 mM DTT (Sigma). Tissues were then minced by scalpel, and were then resuspended in DMEM plus collagenase I for 1 h. Single-cell suspensions were then pelleted and washed, before resuspending the cells in culture medium (DMEM, 15 %-FBS, 2 mM glutamine, 1 mM pyruvate, 10 mM HEPES, 100 units/mL penicillin/streptomycin, 0.1 mg/Ml gentamicin, and 2 g/l fungizone) [13]. The primary cells were labeled passage 1–10. The study was approved by the institutional review board of all authors’ institutions. All clinical investigations were in accordance with the principles expressed in the Declaration of Helsinki.
Methylthiazol tetrazolium assay
Cells were seeded in a 96-well plate at a density of 5000 cells per well. At indicated time points, 15 μL of methylthiazol tetrazolium (MTT) solution per well (5 mg/mL in PBS) was added. After 2 h of incubation, 150 μL of DMSO per well was added to dissolve the crystals. The plate was allowed to stand for 10 min at room temperature, and the absorbance (OD value) at 490 nm was recorded.
Cell death assay
Cell death was estimated by trypan blue staining assay. The number of dead cell was recorded.
Clonogenic assay
One thousand viable PANC-1 cells were plated in 100-mm petri dishes. Medium with or without ST-3595 was refreshed every 2 days. Cells were incubated for a total of 10 days in a humidified incubator containing 5 % CO2 in air at 37 °C. After 4 % paraformaldehyde fixing for 15 min, colonies were stained with 2 % crystal violet and then manually counted.
Apoptosis assay by flow cytometry
The apoptosis was detected by flow cytometry through annexin V-FITC/propidium iodide (PI) staining. Cells were incubated in 6-well plates. After treatment, cells were collected, washed with PBS and resuspended in binding buffer containing Annexin V-FITC and PI according to the manufacturer’s instructions. After staining, cells were analyzed by flow cytometry (Becton-Dickinson, San Jose, CA).
Apoptosis quantification by histone-DNA ELISA assay
Cell death detection enzyme-linked immunosorbent assay (ELISA) kit (Roche, Mannheim, Germany) was utilized to quantify cell apoptosis according to the manufacturer’s protocols.
Detection of mitochondrial membrane potential
The mitochondrial membrane potential (MMP) was measured through JC-10 dye (Invitrogen, Shanghai, China) as reported [14]. After treatment, the staining of the cells was performed by incubation of JC-10 dye in culture medium for 10 min at 37 °C, afterwards, cells were washed, and JC-10 green fluorescence intensity, indicator of MMP reduction (ΔΨm), was measured immediately by a fluorescence microplate reader (Titertek Fluoroscan, Meckenheim, Germany) at 485 nm wavelength with a reference filter of 538 nm.
Western blots
After applied treatment, cells were lysed with lysis buffer containing a protease inhibitor cocktail (P8340, Sigma-Aldrich). Protein mixtures were separated by 10 % SDS-PAGE and further analysis was performed by Western blots. Membranes were incubated overnight with primary antibody, followed by incubation with the horseradish peroxidase-conjugated second-step antibody (Santa Cruz Biotech, Santa Cruz, CA). The results were visualized using the enhanced chemiluminescence substrate kit (ECL kit, Amersham Biosciences, NJ).
Cyp-D siRNA and transfection
Two non-overlapping siRNAs against human Cyp-D (Cyp-D siRNA-1 and Cyp-D siRNA-2) as well as the negative control siRNA were purchased from Dharmacon Research Inc. (Lafayette, CO). We applied siRNA duplexes at a final concentration of 200 nM using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA) for transfection. The transfection took 24 h, and was repeated once for another 24 h. Expression of Cyp-D and tubulin (loading) was tested by Western blots.
HDAC activity assay
HDAC activity was determined using the colorimetric HDAC activity assay kit (Calbiochem, San Diego, CA) according to the manufacturer’s instructions. Briefly, 10 μg of whole-cell extract for each sample were incubated with the HDAC assay buffer and the HDAC colorimetric substrate for 30 min at 37 °C. Lysine developer was then added and the samples were incubated at 37 °C for another 30 min and then read in an ELISA plate reader at 405 nm.
Xenograft assay
Experiments carried out using 7–10-week-old female nude mice (Animal Care Facility, Zhejiang University, Hangzhou, China) were approved by the Ethics Committee for Animal Care according to international guidelines. Exponentially growing PANC-1 cells (2.5 × 106 cells/mouse) were s.c. injected into the right flank (6 mice/group). Treatment started 2 weeks after tumor implant, when tumors reached a volume around 100 mm3. ST-3595 was dissolved in DMSO/ethanol/PBS (10:5:85) and administered orally at 25 mg/kg daily for 2 weeks. Gemcitabine (30 mg/kg) was administered i.v. daily for 2 weeks. Tumor volume was calculated using the formula: TV (mm3) = (d 2 × D)/2, in which d and D were the shortest and the longest diameter, respectively.
Statistical analysis
Statistical analysis was carried out using the SPSS 18.0 software. All values were expressed as the mean ± standard deviation (SD). A P value, calculated by ANOVA, of less than 0.05 was considered statistically significant.
Results
ST-3595 potently inhibits pancreatic cancer cell survival and proliferation
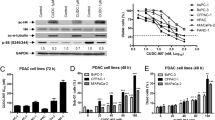
First, we evaluated the potential effect of ST-3595 on pancreatic cancer cell proliferation. MTT assay was performed. As shown in Fig. 1a, ST-3595 dose-dependently decreased the viability of PANC-1 cells, which is an established pancreatic cancer cell line. Further, the results from the trypan blue assay showed that ST-3595 (1–5 μM) induced significant PANC-1 cell death (Fig. 1b). Clonogenicity assay results in Fig. 1c demonstrated that ST-3595 inhibited PANC-1 colony formation, further demonstrating its anti-proliferative activity. Meanwhile, ST-3595 exerted similar effects in two other established pancreatic cancer cell lines (AsPC-1 and Mia-PaCa-2) (Fig. 1d, e). In patient-derived primary pancreatic cancer cells, treatment with ST-3595 again inhibited cell proliferation (Fig. 1f). Notably, ST-3595 at 2.5 μM failed to induce significant cytotoxicity to HPDE6c7 cells (Fig. 1a, b). HPDE6c7 is a non-cancerous pancreatic epithelial cell line [12]. Together, these results show that ST-3595 exerts potent anti-proliferative and cytotoxic effects against established and patient pancreatic cancer cells.

ST-3595 inhibits pancreatic cancer cell proliferation. The viability of PANC-1 cells (a), HPDE6c7 cells (a), AsPC-1 cells (d), Mia-PaCa-2 cells (e), or patient-derived pancreatic cancer cells (f) after applied ST-3595 treatment was tested by MTT assay. Death of PANC-1 cells or HPDE6c7 cells after applied ST-3595 treatment was tested by trypan blue assay (b). Number of PANC-1 colonies with indicated ST-3595 treatment was presented (c), representative colony images were also shown (d, upper panels). Experiments were repeated three times, and similar results were observed. For each assay, n = 5. Data were expressed as mean ± SD (same to all figures). *P < 0.05 (vs. ST-3539 0 μM) was considered statistically significant
ST-3595 induces caspase-dependent apoptosis in pancreatic cancer cells
The effect of ST-3595 on pancreatic cancer cell apoptosis was also tested. Annexin V FACS assay was performed, and results in Fig. 2a showed that ST-3595 treatment dose-dependently increased the number of Annexin V-positive PANC-1 cells, indicating apoptosis activation. Histone-DNA apoptosis ELISA assay results in Fig. 2b further confirmed pro-apoptosis activity by ST-3595 in PANC-1 cells. Similar results were also reproduced in AsPC-1 cells (Fig. 2c) and patient-derived pancreatic cancer cells (Fig. 2d). Notably, in HPDE6c7 cells, ST-3595 at 2.5 μM failed to induce significant apoptosis, as tested by Annexin V FACS assay (Fig. 2a) or histone-DNA apoptosis ELISA assay (Fig. 2b). To investigate the association between apoptosis and ST-3595-induced cytotoxicity, two caspase inhibitors were utilized. Results showed that the pan caspase inhibitor z-VAD-fmk and the caspase-8 inhibitor z-ITED-fmk alleviated ST-3595-induced PANC-1 cell growth inhibition (Fig. 2e) and death (Fig. 2f). Further, the caspase inhibitors showed similar effects in primary cancer cells (Fig. 2g). These results indicate that apoptosis activation mediates ST-3595-induced cytotoxicity in established and patient pancreatic cancer cells.

ST-3595 induces pancreatic cancer cell apoptosis. The apoptosis of PANC-1 cells (a–b), HPDE6c7 cells (a–b), AsPC-1 cells (c), or patient-derived pancreatic cancer cells (d) after applied ST-3595 treatment was tested by the Annexin V FACS assay and/or the histone-DNA apoptosis ELISA assay. The effect of z-VAD-fmk (vad, 50 μM), and z-ITED-fmk (ited, 50 μM) on ST-3595 (2.5 μM, 72 h)-induced pancreatic cancer cell viability reduction or death was tested (e–g). Experiments were repeated three times, and similar results were observed. For each assay, n = 5. *P < 0.05 (vs. ST-3539 0 μM) (a–d). C stands for ST-3539 0 μM (e–g). *P < 0.05 (e–g)
ST-3595-induced pancreatic cancer cell apoptosis is associated with HDAC inhibition and mitochondrial permeability transition pore opening
Next, we explored the underlying mechanisms of ST-3595-induced activities in pancreatic cancer cells. Since ST-3595 is a novel HDAC inhibitor, the activity and expression of HDACs in ST-3595-treated pancreatic cancer cells were tested. As demonstrated, ST-3595 treatment dose-dependently inhibited HDAC activity in both established (Fig. 3a) and patient-derived pancreatic cancer cells (Fig. 3b). In PANC-1 cells, ST-3595 was more efficient than SAHA at the same concentration (2.5 μM) in inhibiting HDAC activity (Fig. 3a). Expressions of HDACs, including HDAC-1, −2, −4, and −6, were not significantly affected by the ST-3595 treatment in the above cancer cells (Fig. 3a, b).

ST-3595 inhibits HDAC activity and induces MPTP opening. The HDAC activity of PANC-1 cells or patient-derived pancreatic cancer cells with indicated ST-3595 or SAHA treatment was shown (a and b), expressions of HDAC-1/-2/-3/-6 were also demonstrated (a and b, upper panels). The MMP level of PANC-1 cells or patient-derived pancreatic cancer cells with applied ST-3595 treatment was presented (c and d). PANC-1 cells or patient-derived pancreatic cancer cells, pretreated with CsA (1 μM, 1 h), SfA (1 μM, 1 h), or Cyp-D siRNA (−1/-2), were then treated with applied ST-3595 for indicated time, cell apoptosis (for PANC-1 cells, e) and viability (for primary cells, f) were presented. Expressions of Cyp-D and tubulin were also shown (e and f, upper panels). Experiments were repeated three times, and similar results were observed. For each assay, n = 5. Data were expressed as mean ± SD. *P < 0.05 (vs. ST-3539 0 μM) (a–d). # P < 0.05 vs. ST-3539 2.5 μM (e and f). C stands for ST-3539 0 μM (e and f)
Many anti-cancer drugs-induced cell apoptosis is associated with mitochondrial permeability transition pore (MPTP) opening [15–17]. Here, we noted that the mitochondrial membrane potential (MMP) level was decreased after indicated ST-3595 treatment in established and patient pancreatic cancer cells, also indicating MPTP opening (Fig. 3c, d). Importantly, MPTP inhibitors, including cyclosporin A (CsA) and sanglifehrin A (SfA), remarkably inhibited ST-3595-induced apoptosis in PANC-1 cells (Fig. 3e) and in primary cancer cells (data not shown). Further, siRNA-mediated downregulation of Cyp-D (Fig. 3e, f), the key component of MPTP [18], also alleviated apoptosis by ST-3595 in PANC-1 (Fig. 3e) and primary cancer cells (data not shown). Note that two non-overlapping siRNAs against Cyp-D were utilized here, and both of them led to dramatic Cyp-D downregulation in transfected cells (Fig. 3e, f). In patient-derived pancreatic cancer cells, CsA, SfA, and Cyp-D siRNAs also suppressed ST-3595-induced growth inhibition (Fig. 3f). These results suggest that ST-3595 inhibits HDAC activity, and induces MPTP-mediated apoptosis in pancreatic cancer cells.
ST-3595 sensitizes gemcitabine-mediated anti-pancreatic cancer activity in vitro and in vivo
Clinically, gemcitabine is the only approved chemotherapeutic agent for pancreatic cancer treatment, showing only limited value in improving patients’ survival [6, 19]. We thus examined the potential effect of ST-3595 on gemcitabine. In PANC-1 cells, gemcitabine (10 μM) treatment alone only induced minor viability reduction and apoptosis (Fig. 4a, b); co-administration with a low concentration of ST-3595 (1 μM) significantly increased gemcitabine’s sensitivity, leading to substantial PANC-1 cell death (Fig. 4a) and apoptosis (Fig. 4b). The gemcitabine-sensitization activity by ST-3595 was also observed in patient pancreatic cancer cells (Fig. 4c). While in non-cancerous epithelial HPDE6c7 cells, ST-3595 and gemcitabine co-administration induced much weaker cytotoxicity and apoptosis (Fig. 4a, b).

ST-3595 and gemcitabine synergism against pancreatic cancer cells. PANC-1 cells (a–b), HPDE6c7 cells (a–b), or patient-derived pancreatic cancer cells (c) were treated with gemcitabine (Gem, 10 μM) and/or ST-3595 (ST, 1 μM) for 72 h (a and c) or 48 h (b), cell viability and/or apoptosis were tested. In vivo growth of PANC-1 xenografts in nude mice administrated with gemcitabine (Gem, 30 mg/kg, i.v. daily, 14 days) and/or ST-3595 (ST, oral administration, 25 mg/kg, daily, 14 days) was shown (d), at the end of experiments, xenografted tumors were isolated and weighted (e). For each in vitro assay, n = 5. Data were expressed as mean ± SD. *P < 0.05. C stands for untreated control (a–d)
The anti-pancreatic cancer activity of ST-3595, alone or in combination with gemcitabine, was also tested in vivo. Results in Fig. 4d showed that the PANC-1 xenograft growth in nude mice was dramatically inhibited by co-administration of gemcitabine and ST-3595. The added strategy was more potent than either single agent in inhibiting PANC-1 cell growth in vivo (Fig. 4d), although ST-3595 alone also inhibited growth of xenografts in nude mice (Fig. 4d). The tumor weights results in Fig. 4e further confirmed in vivo gemcitabine-sensitization effect by ST-3595. Note that the mice body weights were not affected by the single or the combination treatment (data not shown). Together, these results show that ST-3595 sensitizes gemcitabine-mediated anti-pancreatic cancer activity in vitro and in vivo.
Discussions
This current study investigated whether ST-3595, a novel pan-HDAC inhibitor, had an effect against pancreatic cancer cells, either alone or in combination with gemcitabine. Growth inhibition and apoptosis are two key mechanisms by which chemotherapeutic agents induce cytotoxic effects in cancer cells. Our results showed that ST-3595 inhibited proliferation in both established and patient-derived pancreatic cancer cells. Meanwhile, this HDAC inhibitor activated caspase-dependent apoptosis in the above cancer cells. More importantly, it potently potentiated the cytotoxic effect of gemcitabine in pancreatic cancer cells. In addition, our in vivo results demonstrated that ST-3595 administration inhibited growth of PANC-1 xenografts in nude mice, and this activity was further enhanced when used in combination with gemcitabine.
Our results suggest that ST-3595-induced pancreatic cancer cell apoptosis was associated with MPTP opening. The MPTP is a channel complex that is mainly composed of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM), the adenine nucleotide translocator (ANT) located in the inner mitochondrial membrane (IMM), and Cyp-D in the matrix, along with some other molecules [17, 18, 20]. CsA binds to Cyp-D and blocks its peptidyl-prolylcis-transisomerase (PPIase) activity to shut down MPTP [21, 22]. SfA also associates with Cyp-D at a different site, similarly blocking the PPIase activity [21]. Here, we found that both CsA and SfA remarkably inhibited ST-3595-induced pancreatic cancer cell apoptosis, suggesting the involvement of MPTP in ST-3595-induced cell apoptosis. Further, pancreatic cancer cells with siRNA knockdown of Cyp-D were protected from ST-3595. Together, these results indicate that Cyp-D-dependent MPTP opening is required, at least in part, for ST-3595-induced pancreatic cancer cell apoptosis.
Gemcitabine is the gold standard chemotherapeutic drug for the treatment of pancreatic cancer. The response of pancreatic cancers, especially the advanced forms, to gemcitabine-based therapy is still unsatisfactory [2, 5]. An improvement of the disease outcome may be obtained by sensitization of the activity of gemcitabine [2, 5]. The data presented here showed that ST-3595 exerted anti-proliferative and pro-apoptosis activities to pancreatic cancer cells. Further, this novel HDAC inhibitor dramatically sensitized cancer cells to gemcitabine both in vitro and in vivo. These findings indicate that ST-3595 may have a potential as a lead compound for future development of anti-pancreatic cancer therapy.
References
Costello E, Neoptolemos JP. Pancreatic cancer in 2010: new insights for early intervention and detection. Nat Rev Gastroenterol Hepatol. 2011;8:71–3.
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17.
Ducreux M, Boige V, Malka D. Treatment of advanced pancreatic cancer. Semin Oncol. 2007;34:S25–30.
Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703.
Blaszkowsky L. Treatment of advanced and metastatic pancreatic cancer. Front Biosci. 1998;3:E214–25.
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (hdacs): characterization of the classical hdac family. Biochem J. 2003;370:737–49.
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202.
Feng W, Zhang B, Cai D, Zou X. Therapeutic potential of histone deacetylase inhibitors in pancreatic cancer. Cancer Lett. 2014;347:183–90.
Koutsounas I, Giaginis C, Theocharis S. Histone deacetylase inhibitors and pancreatic cancer: are there any promising clinical trials? World J Gastroenterol. 2013;19:1173–81.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Bu HQ, Liu DL, Wei WT, Chen L, Huang H, Li Y, et al. Oridonin induces apoptosis in sw1990 pancreatic cancer cells via p53- and caspase-dependent induction of p38 mapk. Oncol Rep. 2014;31:975–82.
Min H, Xu M, Chen ZR, Zhou JD, Huang M, Zheng K, et al. Bortezomib induces protective autophagy through amp-activated protein kinase activation in cultured pancreatic and colorectal cancer cells. Cancer Chemother Pharmacol. 2014;74:167–76.
Zhen YF, Wang GD, Zhu LQ, Tan SP, Zhang FY, Zhou XZ, et al. P53 dependent mitochondrial permeability transition pore opening is required for dexamethasone-induced death of osteoblasts. J Cell Physiol. 2014;229:1475–83.
Elrod JW, Molkentin JD. Physiologic functions of cyclophilin d and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–22.
Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7.
Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–66.
Javadov S, Kuznetsov A. Mitochondrial permeability transition and cell death: the role of cyclophilin d. Front Physiol. 2013;4:76.
Eckel F, Schneider G, Schmid RM. Pancreatic cancer: a review of recent advances. Expert Opin Investig Drugs. 2006;15:1395–410.
Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–40.
Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin a acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-d at a different site from cyclosporin a. J Biol Chem. 2002;277:34793–9.
Sullivan PG, Thompson MB, Scheff SW. Cyclosporin a attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–34.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Minjie, S., Defei, H., Zhimin, H. et al. Targeting pancreatic cancer cells by a novel hydroxamate-based histone deacetylase (HDAC) inhibitor ST-3595. Tumor Biol. 36, 9015–9022 (2015). https://doi.org/10.1007/s13277-015-3537-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3537-5