
Abstract
A possible association between multiple drug resistance 1 gene (MDR1) polymorphisms and the risk of developing hepatocellular carcinoma (HCC) is currently under debate, and evidence from various epidemiological studies has yielded controversial results. To derive a more precise estimation of the association between MDR1 polymorphisms and HCC risk, the present meta-analysis was performed. A total of 8 studies containing 11 cohorts with 4407 cases and 4436 controls were included by systematic literature search of EMBASE, PubMed, Web of Science, and CNKI. All polymorphisms were classified as mutant/wild-type alleles. In particular, the variation type, functional impact, and protein domain location of the polymorphisms were assessed and used as stratified indicators. The pooled odds ratio (OR) with 95 % confidence interval (CI) was calculated to evaluate the association. Overall, our results suggested that the mutant alleles of the MDR1 gene were associated with a significantly increased risk for HCC under all genetic models (allelic model: OR = 1.28, 95 % CI = 1.20–1.36, P < 0.001; dominant model: OR = 1.27, 95 % CI = 1.16–1.38, P < 0.001; recessive model: OR = 1.59, 95 % CI = 1.36–1.85, P < 0.001). Furthermore, increased risks for HCC were also revealed in stratified analyses by ethnicity, sample size, and quality scores of cohorts as well as variation type, functional impact, and protein domain location of polymorphisms. In conclusion, the present meta-analysis suggested that the presence of MDR1 mutant alleles might be a risk factor for HCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) ranks as the fifth most frequent cancer and the second leading cause of cancer death globally [1]. In China, it is the second most common cancer, and Chinese cases represent half of the total HCC cases worldwide [2]. The high incidence of Chinese HCC is mainly attributed to the prevalence of hepatitis virus infection, especially hepatitis B virus (HBV). HBV infection and subsequent persistent inflammation result in the sequential development of chronic hepatitis, cirrhosis, and HCC [3]. Moreover, etiological studies have implicated that many host factors (including gender, age, and ethnicity), viral factors (including viral genotypes and replication levels), and environmental factors (including aflatoxin exposure, alcohol drinking, and tobacco smoking) are involved in HCC susceptibility [4, 5]. In addition, there is increasing evidence indicating the association between genetic variants and the risk of HCC [6–8]. Among all the genetic factors, the single nucleotide polymorphisms (SNPs) are the most widely studied genetic variations.
Chronic inflammation caused by potential risk factors may result in hepatic accumulation of oxidative and other genotoxic products, possibly leading to the initiation of multistage hepatocarcinogenesis [9–11]. Biliary and sinusoidal clearance of such endogenous and exogenous carcinogenic substances is one of the most important protective functions of the liver. This function is largely achieved by a drug efflux transporter system that comprises mainly ATP-binding cassette transporter (ABC transporter) proteins [12, 13]. Therefore, polymorphisms of these proteins might compromise the efflux of mutagenic factors that could contribute to HCC.
Multiple drug resistance 1 gene (MDR1)-encoded P-glycoprotein (P-gp), one of the most important ABC transporters, is widely expressed in normal cells of various organs, including the liver, kidney, and brain [14–16]. MDR1 is extensively involved in the absorption and elimination of various harmful substances and also participates in regulating cell growth, differentiation, and death [17–19]. Of note, studies have indicated that SNPs in the MDR1 gene have an important impact on the expression and function of P-gp, therefore influencing susceptibility to numerous diseases, including HCC [20–26]. To date, more than 50 SNPs in the MDR1 gene have been documented [27, 28], and an association between these SNPs and the risk of HCC has been proposed; however, contradictory results have been reported and studies focused on one particular polymorphism are also limited [29–36]. Significant correlations of MDR1 polymorphisms and HCC risk were observed in several studies, whereas no significant associations were detected in others [29–36]. Therefore, we included all studies on association between MDR1 SNPs and the risk for HCC and classified the included SNPs as mutant/wild type. To precisely estimate the association of MDR1 mutant/wild-type alleles with HCC susceptibility, we performed the first meta-analysis on all eligible studies.
Materials and methods
Literature search strategy and selection criteria
We performed a systematic review and meta-analysis in accordance with the guidelines of PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement [37] (Additional file 1). We searched PubMed, Web of Science, EMBASE, and CNKI without language limitations till December 1, 2014. The following search algorithm was used: (“hepatocellular carcinoma” or “hepatocellular neoplasm” or “liver cancer” or “HCC”) and (“multiple drug resistance 1 gene” or “MDR1” or “ABCB1” or “P-glycoprotein” or “P-pg”) and (“polymorphism” or “variation” or “susceptibility”). Additionally, all the references cited in the included articles were hand-searched to identify potentially eligible studies.
Inclusion and exclusion criteria
We included studies that (i) evaluated MDR1 gene polymorphisms and HCC risk, (ii) designed as case-control studies, and (iii) provided data for calculating odds ratios (ORs) with corresponding 95 % confidence intervals (CIs). Studies were excluded if (i) no specific controls have been selected, like review, comment articles, and case-only studies; (ii) sufficient data to estimate ORs with 95 % CIs is lacking; and (iii) there is duplication of previous publications or samples.
Data extraction
Two reviewers (Z.C. W and L.Z. L) independently carried out the data extraction process using a predefined standard form. Disagreements were discussed and resolved with consensus. The following information were collected: first author’s name, year of publication, ethnicity, number of patients and controls, age, gender, genotyping method, polymorphism site, exon location, variation type, amino acid alteration, and genotype frequency of cases and controls, respectively. All polymorphisms were classified as mutant/wild-type alleles to unify the statistical analyses.
When the mutant alleles of polymorphism site lead to a nonsynonymous change in the amino acid sequence, functional impacts of corresponding amino acid changes were estimated using online MutationAssessor [38]. The functional impacts were evaluated as neutral, low, or medium impacts according to the estimated potential impacts on the biological activity of MDR1. The locations of altered amino acids in the MDR1 structure were also assessed according to Uniprot.org (http://www.uniprot.org/uniprot/P08183; see details in Additional file 2). Allele frequencies were calculated from the corresponding genotype distributions if not given directly. For the studies that investigated different polymorphisms in one cohort [30, 32, 34], the datasets were recognized as independent studies. Study quality was assessed independently by Z.C. W. and L.Z. L using the 10-point scoring scale for quality of genetic association studies proposed by Clark and Baudouin [39].
Statistical methods
We estimated the HCC risk associated with mutant alleles (using 2 as a representative example) at different polymorphism sites compared with the wild-type alleles (using 1 as a representative example) under the following genotypic models: allele (2 vs. 1); co-dominant (2/2 vs. 1/1, 2/2 vs. 1/2, and 1/2 vs. 1/1); dominant (2/2 + 1/2 vs. 1/1); and recessive (2/2 vs. 1/2 + 1/1). Pooled ORs with corresponding 95 % CIs were calculated to assess the strength of associations between the MDR1 polymorphisms and HCC risk. A P value below 0.05 was considered statistically significant.
Q tests and I 2 tests were performed to test the heterogeneity (indicated large heterogeneity) [40]. A fixed effects model was used when there was no heterogeneity (P ≥ 0.10 and I 2 ≤ 50 %); otherwise (P < 0.10 and I 2 > 50 %), a random effects model was used [41]. Hardy-Weinberg equilibrium (HWE) in controls of each cohort was assessed using the online HWE calculator (http://ihg.gsf.de/cgi-bin/hw/hwa1.pl) (P < 0.05 indicated significant disequilibrium).
In addition, the sources of heterogeneity were investigated by subgroup analysis based on the following: ethnicity, sample sizes (no. of cases ≤1000 or >1000), quality scores (scores >7 or ≤7), variation type (synonymous or nonsynonymous), functional impact (neutral/low or medium), positions in coding sequences (cytoplasmic or transmembrane), and positions in the functional protein domains (ABC transmembrane type 1 or ABC transporter). Sensitivity analyses were performed by deletion of a single dataset each time to assess the impact of the individual dataset to the pooled OR. Begg’s funnel plots [42] and Egger’s linear regression tests [43] were performed to evaluate publication bias. All P values were two-sided, and P < 0.05 was considered statistically significant. Statistical analyses were conducted using Validating Review Manager Version 5.1 (Copenhagen, The Nordic Cochrane Centre, The Cochrane Collaboration, 2011) and STATA 11.0 (StataCorp LP, College Station, TX, USA).
Results
Characteristics of the studies and polymorphisms
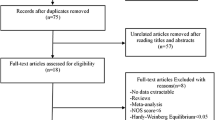
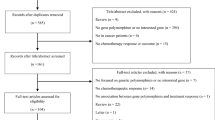
The database search yielded 81 relevant studies. After excluding 73 studies according to the exclusion criteria (Additional file 3), a total of 8 studies that consisted of 11 cohorts with 4407 cases and 4436 controls [29–36] was enrolled. The characteristics of the included studies are shown in Table 1. All 11 cohorts selected healthy individuals as control, and all cohorts were consistent with the Hardy-Weinberg equilibrium except for Minoru F-2 cohort focused on the 3435C > T polymorphism of MDR1 [30].
Furthermore, different polymorphisms of the included studies are listed in Table 2 with exonic location. All polymorphisms were classified as mutant/wild-type alleles. In particular, the corresponding variation type, amino acid alteration with related functional impact, feature key, calculated positions in the coding sequence, and positions in the protein functional domain are also shown (Table 2).
Meta-analysis
In the overall analyses of all 11 cohorts, a significant association was detected under all genetic models, suggesting that the mutant alleles were associated with an increased HCC risk (Fig. 1 and Table 3): (i) allelic model, 2 vs. 1: OR = 1.28, 95 % CI 1.20–1.36, P < 0.001; (ii) dominant model, 2/2 + 1/2 vs. 1/1: OR = 1.27, 95 % CI 1.16–1.38, P < 0.001; (iii) co-dominant model, 2/2 vs. 1/1: OR = 1.81, 95 % CI 1.58–2.07, P < 0.001; (iv) co-dominant model, 2/2 vs. 1/2: OR = 1.49, 95 % CI 1.30–1.70, P < 0.001; (v) co-dominant model, 1/2 vs. 1/1: OR = 1.15, 95 % CI 1.05–1.26, P = 0.002; and (vi) recessive model, 2/2 vs. 1/2 + 1/1: OR = 1.59, 95 % CI 1.36–1.85, P < 0.001. No significant heterogeneity among studies was observed.

Forest plot presenting the meta-analysis of MDR1 polymorphisms and the susceptibility to hepatocellular carcinoma under the allelic model in all cohorts. The horizontal lines represent 95 % confidence intervals for estimating the outcome of the mutant allele versus the wild-type allele in the meta-analysis. Blue squares denote overall estimates of the effects
Subgroup analyses
Subgroup analyses were performed by stratifying of ethnicity, sample size, quality score, variation type, functional impact, position in the coding sequence, and protein functional domain. Of the 11 cohorts, there were only 2 datasets for the Japanese subgroup [30], medium functional impact subgroup [32, 34], and positions in the transmembrane region subgroup [32, 34], respectively. Therefore meta-analyses were not performed in these subgroups.
In the subgroup analyses by ethnicity, a significant association was observed in the Chinese subgroup under the allelic model (OR = 1.28, 95 % CI 1.20–1.36, P < 0.00001; Table 4). Though meta-analyses could not be conducted in the Japanese subgroup, it was found that neither dataset exhibited significant results (Minoru F-1, 2010: OR = 1.23, 95 % CI 0.74, 2.07; P = 0.43; Minoru F-2, 2010: OR = 1.61, 95 % CI 0.95, 2.72; P = 0.72). Likewise, subgroup analyses were performed according to the sample size and quality score and the results indicated significantly increased risks for HCC irrespective of size and quality (Table 4).
In addition to the typical standards used above, we classified these cohorts by the variation type, impacts on protein function, and positions in the coding sequence of MDR1 polymorphisms. In the subgroup analyses of variation type, both synonymous and nonsynonymous subgroups revealed significant results for increasing HCC risk. Interestingly, larger ORs were observed in the nonsynonymous subgroup compared with the synonymous subgroup (nonsynonymous subgroup: OR = 1.31, 95 % CI 1.21–1.41, P < 0.00001; synonymous subgroup: OR = 1.22, 95 % CI 1.08–1.37, P = 0.001). Subgroup analyses according to the estimated functional impacts (FI) indicated that even neutral/low FI resulted in a significantly higher risk for HCC (neutral/low FI subgroup: OR = 1.23, 95 % CI 1.12–1.35, P < 0.00001). However, the cohorts with a medium FI revealed higher ORs (Gao J-2, 2013: OR = 1.65, 95 % CI 1.32–2.05, P < 0.0001; Li XF, 2013: OR = 1.33, 95 % CI 1.13–1.57, P < 0.001). Positions in different coding sequence subgroup analyses revealed that cytoplasmic polymorphisms correlated with a significantly higher HCC risk (cytoplasmic subgroup: OR = 1.28, 95 % CI 1.19–1.37; P < 0.00001), whereas transmembrane polymorphisms exhibited site-specific results (Gao J-2, 2013: OR = 1.65, 95 % CI 1.32–2.05, P < 0.0001; Yang D-1, 2013: OR = 1.65, 95 % CI 0.98–1.33, P = 0.10). Positions in different protein functional domain subgroup analyses indicated that only polymorphisms in the ABC transporter domain rather than the ABC transmembrane type 1 domain significantly increased HCC risk (ABC transporter subgroup: OR = 1.28, 95 % CI 1.18–1.38, P < 0.0001; ABC transmembrane type 1 subgroup: OR = 1.21, 95 % CI 0.98–1.49, P = 0.07). The results indicated that polymorphisms positioned in the cytoplasmic coding sequence and ABC transporter domain play a vital role in hepatocarcinogenesis.
Sensitivity analyses and publication bias
One cohort was excluded at each time to assess the influence of the individual dataset on the overall results. The overall significance was not altered when any single cohort was deleted, suggesting that the results were robust. Begg’s funnel plots and Egger’s tests were conducted to assess the publication bias for the available datasets. The funnel plots were symmetrical (Fig. 2), indicating that there were no publication biases in the studies of MDR1 polymorphisms (Egger’s test, P = 0.640).

Begg’s funnel plot of pseudo 95 % confidence limits. Evaluation of publication bias for the association of MDR1 polymorphisms with hepatocellular carcinoma risk in all cohorts. OR odds ratio, s.e. standard error
Discussion
HCC is a common malignancy resulting from a complex interaction between environmental and genetic factors. With high interest in gene susceptibility to carcinogenesis, increasing efforts have been devoted to the study of genetic variants and HCC risk. Human MDR1 is physiologically widely expressed in the liver, kidney, colon, adrenal gland, and blood-brain barrier [28]. MDR1 plays an important role in protecting organs from xenobiotics or toxins, which can cause chronic inflammation that is thought to be an etiological factor for many diseases, including HCC. Several studies have digged into the association between MDR1 polymorphisms and HCC risk; however, controversial results exist. Hence, we performed the first meta-analysis to clarify the inconsistency among available studies. This meta-analysis included 8 studies (11 cohorts) with 4407 HCC patients and 4436 controls. The sample size is nearly ten times that of the largest cohort included.
Consistent with the biological function of MDR1, wild-type MDR1 was found to be protective against HCC development [27]. The positive association of a mutant allele in MDR1 polymorphisms among all cohorts was detected under allelic and all genotypic models, suggesting that mutant alleles of the MDR1 gene significantly increased HCC risk. In the Chinese subgroup, a positive association of a mutant allele and increased HCC risk was also observed. Meanwhile, subgroup analyses revealed a significant association in both cohorts with large and small sample sizes. However, the I 2 for a large sample size cohort (3 %) was substantially smaller than the I 2 for a small sample size cohort (49 %). Likewise, the I 2 for the high-quality score cohorts and low-quality score cohorts exhibited the same trend as the large/small sample size cohorts. These results indicated that the heterogeneity might be caused by small sample sizes and low-quality scores.
Unlike traditional meta-analysis for gene polymorphisms, the present meta-analysis analyzed multiple polymorphism sites classified as mutant or wild type to explore impacts of MDR1 on HCC risk as a whole, and we included variation type, estimated functional impact, positions in the coding sequence, and the protein functional domain as subgroup stratifiers to assess their site-related influences on HCC risk. In the variation type subgroup analyses, both synonymous and nonsynonymous cohorts indicated that mutant alleles significantly correlated with HCC risk. The effect of nonsynonymous variations was largely due to the alteration of protein structures leading to functional changes in MDR1, thereby causing toxin accumulation in hepatocytes. In contrast, the effect of synonymous variations may partly be due to impacts on transcriptional and translational processes [44, 45]. Using online MutationAssessor, we calculated the estimated functional impact of different SNPs, and subsequent functional impact subgroup analyses indicated that even neutral/low functional impacts had significant results. A medium functional impact may also have significant findings based on individual datasets. Whether the medium functional impact subgroup exhibited greater significance could not be assessed because of the limited data available. Further investigations on medium functional impact SNPs will be of great value. In protein structure location-based subgroup analyses, we found that cytoplasmic variations significantly increased HCC risk. The transmembrane subgroup only contained two cohorts, making them unsuitable for meta-analysis. Interestingly, we noted that the two cohorts that investigated transmembrane SNPs reported contradictory results, and the larger sample size one reached a negative conclusion. Therefore, we cautiously conclude that SNPs in the cytoplasmic region, rather than in the transmembrane region of MDR1, exhibit a significant association with increased HCC susceptibility. A possible explanation is that the cytoplasmic region exhibits multiple biological activity domains that are responsible for downstream signaling pathway activation, whereas the transmembrane region is only an anchor for MDR1 and appears to be less important. This hypothesis was further validated by protein functional domain subgroup analyses, which indicated that the ABC transporter subgroup exhibits a significant association (P < 0.00001), whereas the ABC transmembrane type 1 subgroup exhibits no association with HCC risk.
There are some limitations of our meta-analysis that should be noticed in interpreting the results. First, the ethnicity of the majority of cohorts was Chinese. There were insufficient data for a meta-analysis of Japanese subgroups. Considering that the SNPs in different populations varied with respect to both sites and frequency, we must be cautious in our conclusions. Second, some potential confounding factors that might have biased this finding could not be assessed due to insufficient data, such as age, tobacco smoking, alcohol drinking, and medicine usage. Third, MDR1 possesses at least 50 different SNPs, but sufficient data were available for only 10 SNPs in the present meta-analysis and the impact of a specific SNP on HCC risk cannot be assessed using available data. At least 40 different SNPs and more studies focused on a specific SNP in a larger HCC cohort require further investigation with respect to their association with HCC risk.
In conclusion, this study is the first meta-analysis to summarize the association between MDR1 gene polymorphisms and HCC susceptibility. The results indicated that mutant alleles of MDR1 significantly increase HCC risk. Interestingly, we also, for the first time, revealed that only the SNPs in the cytoplasmic region significantly correlated with increased HCC risk, which requires further experimental confirmation. Further large studies with multi-ethnic groups, high-quality scores, and different ethnic populations will be necessary to further validate our conclusions.
References
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–55. doi:10.1016/S0140-6736(11)61347-0.
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi:10.3322/caac.21208.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76. doi:10.1053/j.gastro.2007.04.061.
Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558–67. doi:10.1016/S1473-3099(05)70216-4.
Tanaka M, Katayama F, Kato H, Tanaka H, Wang J, et al. Hepatitis B and C virus infection and hepatocellular carcinoma in China: a review of epidemiology and control measures. J Epidemiol. 2011;21(6):401–16.
Wang ZC, Gao Q, Shi JY, Yang LX, Zhou J, et al. Genetic polymorphism of the kinesin-like protein KIF1B gene and the risk of hepatocellular carcinoma. PLoS One. 2013;8(4):e62571. doi:10.1371/journal.pone.0062571.
Dragani TA. Risk of HCC: genetic heterogeneity and complex genetics. J Hepatol. 2010;52(2):252–7. doi:10.1016/j.jhep.2009.11.015.
Jiang DK, Sun J, Cao G, Liu Y, Lin D, et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet. 2013;45(1):72–5. doi:10.1038/ng.2483.
Park EJ, Lee JH, Yu GY, He G, Ali SR, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi:10.1016/j.cell.2009.12.052.
Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56(3):704–13. doi:10.1016/j.jhep.2011.09.020.
Aleksandrova K, Boeing H, Nothlings U, Jenab M, Fedirko V, et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology. 2014. doi:10.1002/hep.27016.
Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92. doi:10.1146/annurev.biochem.71.102301.093055.
Szakacs G, Varadi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov Today. 2008;13(9-10):379–93. doi:10.1016/j.drudis.2007.12.010.
Ros JE, Libbrecht L, Geuken M, Jansen PL, Roskams TA. High expression of MDR1, MRP1, and MRP3 in the hepatic progenitor cell compartment and hepatocytes in severe human liver disease. J Pathol. 2003;200(5):553–60. doi:10.1002/path.1379.
Ernest S, Rajaraman S, Megyesi J, Bello-Reuss EN. Expression of MDR1 (multidrug resistance) gene and its protein in normal human kidney. Nephron. 1997;77(3):284–9.
Marchi N, Hallene KL, Kight KM, Cucullo L, Moddel G, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med. 2004;2:37. doi:10.1186/1741-7015-2-37.
Enokida H, Shiina H, Igawa M, Ogishima T, Kawakami T, et al. CpG hypermethylation of MDR1 gene contributes to the pathogenesis and progression of human prostate cancer. Cancer Res. 2004;64(17):5956–62. doi:10.1158/0008-5472.CAN-04-0081.
Schaich M, Ritter M, Illmer T, Lisske P, Thiede C, et al. Mutations in ras proto-oncogenes are associated with lower mdr1 gene expression in adult acute myeloid leukaemia. Br J Haematol. 2001;112(2):300–7.
Batetta B, Mulas MF, Petruzzo P, Putzolu M, Bonatesta RR, et al. Opposite pattern of MDR1 and caveolin-1 gene expression in human atherosclerotic lesions and proliferating human smooth muscle cells. Cell Mol Life Sci. 2001;58(8):1113–20.
Jamroziak K, Mlynarski W, Balcerczak E, Mistygacz M, Trelinska J, et al. Functional C3435T polymorphism of MDR1 gene: an impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur J Haematol. 2004;72(5):314–21. doi:10.1111/j.1600-0609.2004.00228.x.
Kurzawski M, Drozdzik M, Suchy J, Kurzawski G, Bialecka M, et al. Polymorphism in the P-glycoprotein drug transporter MDR1 gene in colon cancer patients. Eur J Clin Pharmacol. 2005;61(5-6):389–94. doi:10.1007/s00228-005-0926-5.
Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer. 2003;10(1):43–73.
Sohn JW, Lee SY, Lee SJ, Kim EJ, Cha SI, et al. MDR1 polymorphisms predict the response to etoposide-cisplatin combination chemotherapy in small cell lung cancer. Jpn J Clin Oncol. 2006;36(3):137–41. doi:10.1093/jjco/hyi231.
Taniguchi S, Mochida Y, Uchiumi T, Tahira T, Hayashi K, et al. Genetic polymorphism at the 5′ regulatory region of multidrug resistance 1 (MDR1) and its association with interindividual variation of expression level in the colon. Mol Cancer Ther. 2003;2(12):1351–9.
Wu L, Xu X, Shen J, Xie H, Yu S, et al. MDR1 gene polymorphisms and risk of recurrence in patients with hepatocellular carcinoma after liver transplantation. J Surg Oncol. 2007;96(1):62–8. doi:10.1002/jso.20774.
Yu X, Xie H, Wei B, Zhang M, Wang W, et al. Association of MDR1 gene SNPs and haplotypes with the tacrolimus dose requirements in Han Chinese liver transplant recipients. PLoS One. 2011;6(11):e25933. doi:10.1371/journal.pone.0025933.
Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75(1):13–33. doi:10.1016/j.clpt.2003.09.012.
Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22(47):7468–85. doi:10.1038/sj.onc.1206948.
Chen Y-D, Yang F, Feng S-T, et al. A case-control study on the association between genetic polymorphisms of MDR1 and hepatic cell cancer susceptibility. Chin Clin Oncol. 2009;14(12):1007–81.
Fukuda MKY, Hirota T, et al. Genetic polymorphisms of hepatic ABC-transporter in patients with hepatocellular carcinoma. JCT. 2010;1:114–23.
Ren YQ, Han JQ, Cao JB, Li SX, Fan GR. Association of MDR1 gene polymorphisms with susceptibility to hepatocellular carcinoma in the Chinese population. Asian Pac J Cancer Prev. 2012;13(11):5451–4.
Gao J. Association of MDR1 gene polymorphisms with the risk of hepatocellular carcinoma in the Chinese Han population. Braz J Med Biol Res. 2013;46(3):311–7.
Jing Rui DX, Deng W, et al. Correlation between MDR1 polymorphism and primary liver cancer in Guangxi. Chin J Oncol Prev Treat. 2013;5(2):122–5.
Yang D, Zhou F, Wang X, Gao H, Li G, et al. Association analysis between MDR1 gene polymorphisms and risk of hepatocellular carcinoma in Chinese population. Biomarkers. 2013;18(3):236–41. doi:10.3109/1354750X.2013.773079.
Li XF, He HB, Zhu YS, He JK, Ye WW, et al. Association between the c.3751G > a genetic variant of MDR1 and hepatocellular carcinoma risk in a Chinese Han population. Asian Pac J Cancer Prev. 2013;14(9):5361–5.
Wan YY, Wang XW, Hui HX, Wan L. Association between the c.1564A > T genetic polymorphism of the MDR1 gene and hepatocellular carcinoma in Chinese population. Genet Mol Res. 2014;13(3):6820–6. doi:10.4238/2014.August.29.3.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013;14 Suppl 3:S7. doi:10.1186/1471-2164-14-S3-S7.
Clark MF, Baudouin SV. A systematic review of the quality of genetic association studies in human sepsis. Intensive Care Med. 2006;32(11):1706–12. doi:10.1007/s00134-006-0327-y.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–88.
DerSimonian R. Combining evidence from clinical trials. Anesth Analg. 1990;70(5):475–6.
Lau J, Ioannidis JP, Terrin N, Schmid CH, Olkin I. The case of the misleading funnel plot. BMJ. 2006;333(7568):597–600. doi:10.1136/bmj.333.7568.597.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–34.
Pagani F, Raponi M, Baralle FE. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc Natl Acad Sci U S A. 2005;102(18):6368–72. doi:10.1073/pnas.0502288102.
Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12(10):683–91. doi:10.1038/nrg3051.
Conflicts of interest
None
Financial support
This work was supported by the Major Program of NSFC (No. 81030038), National Key Sci-Tech Project (2012ZX10002011-002), National Natural Science Foundation of China (Nos. 81372648, 81272730, 81272725), FANEDD (No. 201183), and Shanghai “Promising Youth Medical Worker” Project (No. 13Y055).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhi-Chao Wang, Long-Zi Liu and Xin-Yang Liu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1
PRISMA 2009 checklist of this systematic review and meta-analysis. (DOCX 32 kb)
Additional file 2
MDR1 protein sequence annotation from Uniprot website. (DOCX 108 kb)
Additional file 3
Selection of the related studies. (GIF 46 kb)
Additional file 4
List of included studies. (DOCX 106 kb)
Rights and permissions
About this article

Cite this article
Wang, ZC., Liu, LZ., Liu, XY. et al. Genetic polymorphisms of the multidrug resistance 1 gene MDR1 and the risk of hepatocellular carcinoma. Tumor Biol. 36, 7007–7015 (2015). https://doi.org/10.1007/s13277-015-3407-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3407-1