
Abstract
Fenofibrate, a fibric acid derivative, is known to possess lipid-lowering effects. Although fenofibrate-induced peroxisome proliferator-activated receptor alpha (PPARα) transcriptional activity has been reported to exhibit anticancer effects, the underlying mechanisms are poorly understood. In this study, we investigated the mechanisms behind the antiproliferative effects of fenofibrate in U87MG cells (human glioma cell line) using the WST-8 Cell Proliferation Assay Kit. Furthermore, we examined genome-wide gene expression profiles and molecular networks using the DAVID online software. Fenofibrate reduced the expression of 405 genes and increased the expression of 2280 genes. DAVID analysis suggested that fenofibrate significantly affected cell cycle progression and pathways involved in cancer, including the mTOR signaling pathway and insulin signaling pathway. Results of flow cytometry analysis indicated that fenofibrate induced cell cycle G0/G1 arrest in U87MG cells. Furthermore, we identified the FoxO1–p27kip signaling axis to be involved in fenofibrate-induced cell cycle arrest. Our findings suggest that in addition to its known lipid-lowering effects, fenofibrate may be used as an antitumor agent in glioma therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most common and lethal cancer of the central nervous system. However, conventional therapies, including surgical resection, followed by radiation and chemotherapy, do not significantly improve patient outcomes [1, 2]. Therefore, there is an urgent need to investigate the molecular pathways involved in the development and progression of glioma.
Fenofibrate (FF) is a potent ligand of peroxisome proliferator-activated receptor alpha (PPARα) and has been historically used to regulate glucose and lipid metabolism for treating different forms of hyperlipidemia and hypercholesterolemia [3]. In recent years, FF has been shown to exert interesting anticancer effects. Jiao reported that FF inhibits the growth of human HepG2 cells in a dose-related manner via PPARα-dependent mechanisms [4]. Its other PPARα-dependent effects include inhibition of endothelial cancer cell growth [5], inhibition of tumor angiogenesis [6], and inhibition of glioblastoma and melanoma cell movement and metastases [7, 8]. Previous work suggest that FF may be an effective treatment alternative, not only because of its antiproliferative effects but also because of its low toxicity [9, 10].
FoxO1 is one of the Fork-head box O (FoxO) transcription factors, which are involved in the development of various types of human cancers, including breast carcinomas [11], endometrial cancer [12], prostate cancer [13], and renal carcinomas [14]. Moreover, as a substrate of Akt, FoxO1 plays a causal role in tumor suppression and results in the transcriptional activation of p27kip [15, 16]. A recent study has shown that targeting FoxO1 could effectively induce glioma cell death and inhibit tumor growth by controlling the expression of cell cycle-related genes [17].
Here we report that as a PPARα ligand, FF induces cell cycle arrest at the G0/G1 phase and inhibits the growth of human glioma U87MG cells in a PPARα-dependent manner. We also confirmed that these anticancer effects of FF are mediated via the PPARα/FoxO1/p27kip pathway.
Materials and methods
Human tissue samples and cell culture
Messenger RNA (mRNA) expression data for 89 GBM patients was downloaded from the Chinese Glioma Genome Atlas (CGGA) data portal (http://www.cgga.org.cn/portal.phpg). The TCGA mRNA expression microarray data for 169 GBM patients were downloaded from the following portal: http://tcga-data.nci.nih.gov/tcga/homepage.htm. U87MG cells were purchased from the Chinese Academy of Sciences Cell Bank and grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS).
Oligonucleotides and transfection
PPARα small interfering RNA (siPPARα) was synthesized by GenePharma (Shanghai, China). The following sequences were used for siPPARα: sense, 5′-UCA CGG AGC UCA CAG AAU UUU-3′; antisense: 3′-AAU UCU GUG AGC UCC GUG AUU-5′. FoxO1 small interfering RNA (siFoxO1) was obtained from Cell Signaling Technology (CST, USA). An siRNA that was unrelated to any human sequence was used as a negative control. For transfection, oligonucleotides were complexed with Lipofectamine 2000 using OPTI-MEM (Invitrogen, USA) and added to U87MG cells at a final concentration of 100 nM, following which cells were incubated for 6–8 h at 37 °C before changing the medium.
WST-8 growth assay
U87MG cells were seeded in 96-well culture plates at 2000 cells/well/100 μL. The cells were treated with 0, 50, 100, or 150 μM FF for 1–4 days. Then, tetrazolium monosodium salt WST-8 (Dojindo, Japan) was added (10 μL/well). After incubation for 2 h, the absorbance was determined using a microplate reader (Bio-Rad, USA) at 450-nm wavelength with the reference wavelength set at 630 nm.
Colony formation assay
In total, 5 × 102 U87MG cells/plate were placed onto separate 60-mm tissue culture plates to which different concentrations of FF were added (0, 50, or 100 μM). After incubation for 2 weeks, visible colonies were fixed with 4 % methanol for 30 min and then stained with 0.1 % crystal violet for 20 min. Colony-forming efficiency was determined as the number of colonies/plated cells × 100 %.
Whole genome gene profile chip
Untreated U87MG cells or cells treated with FF (100 μM) for 48 h were harvested, after which total RNA was isolated using the RNeasy Kit (Qiagen, Germany), according to the manufacturer’s instructions. RNA concentration and quality were measured using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA). Four micrograms of total RNA were subjected to mRNA sequencing library preparation before they were sequenced using the Illumina GAII platform (Illumina Inc, USA) according to the manufacturer’s instructions (BGI-Shenzhen, China). The expression fold change (treated cells versus untreated cells) for each gene was calculated as the log2 ratio using the normalized TPM (transcripts per million reads) values. Sets of differentially expressed genes were clustered into biological modules on the basis of gene ontology using Functional Annotation Bioinformatics Microarray Analysis (DAVID) online software (http://david.abcc.ncifcrf.gov).
PPARα transcription factor assay
After FF treatment, nuclear proteins were extracted using the Nuclear/Cytoplasmic Fractionation Kit (KenGEN, China). Equal amounts of protein (20 μg/well) were used to determine PPARα transcriptional activity using the PPARα Transcription Factor Assay Kit (Abcam, USA) according to the manufacturer’s instructions. The absorbance was determined using a microplate reader (Bio-Rad, USA) at 450-nm wavelength.
Cell cycle analysis
After FF treatment, U87MG cells were collected and fixed with 70 % ethanol for at least 12 h at −20 °C. DNA was stained by incubating cells in 50 mg/mL propidium iodide (PI) (Sigma-Aldrich, USA) and 20 g/mL RNase A (Boehringer-Mannheim, Germany) at room temperature for 1 h. The cells were then analyzed by FACScan (Becton-Dickinson, USA).
Western blot analysis
Cells were washed twice with PBS, and proteins were extracted after lysing cells in RIPA lysis buffer (KenGEN, China). Subcellular fractionation was conducted to separate cytoplasmic and nuclear proteins using the Nuclear/Cytoplasmic Fractionation Kit (KenGEN, China). Equal amounts of protein were separated by 10 or 12 % SDS–PAGE, followed by electrotransfer onto polyvinylidene difluoride membranes (Thermo Scientific, USA). Membranes were blocked with 5 % nonfat milk at room temperature for 2 h and then incubated with primary antibodies at 4 °C overnight. Blots were washed three times with 1× TBST (10 min each wash) and then incubated with a secondary antibody (antirabbit or antimouse immunoglobulins) at 1:5000 dilution for 2 h. Membranes were developed using an enhanced chemiluminescence detection system (GE Healthcare, England). The antigen was then visualized using ChemiDoc XRS+ gel imaging system (Bio-Rad, USA), and data were analyzed using ImageJ software. The following primary antibodies were used for immunoblot analysis: PPARα (1:1000; Abcam, USA); p27kip (1:500; Santa Cruz, USA); FoxO1 (1:1000; CST, USA); HDAC1 (1:1000; CST, USA), and β-actin (1:1000; CST, USA).
Statistical analysis
Results are given as mean ± standard deviation (SD) of at least three independent experiments. Statistical comparisons were made using one-way analysis of variance (ANOVA) and Student’s t test (two-tailed) using the SPSS 13.0 software package. P < 0.5 were considered statistically significant.
Results
FF inhibits U87MG cell proliferation
To investigate the effects of FF on U87MG cell proliferation, U87MG cells were treated with different doses of FF (0, 50, 100, and 150 μM) for 1, 2, 3, and 4 days. Proliferation of these FF-treated cancer cells was then assessed using WST-8 assays. As shown in Fig. 1a, FF significantly inhibited cell proliferation in a time- and dose-dependent manner. In addition, treating cells with 100 μM FF for 48 h reduced cell viability by 54.62 ± 1.56 %. FF treatment (50 and 100 μM) also resulted in complete inhibition of U87MG clonogenic growth (Fig. 1b), which further supported its antiproliferative effects.

Human glioblastoma U87MG cell proliferation is inhibited by fenofibrate. a WST-8 assays were conducted on U87MG cells after treatment with fenofibrate at different concentrations (0, 50, 100, and 150 μM). b Colony formation assay was performed on U87MG cells after treatment with fenofibrate at different concentrations (0, 50, and 100 μM). **P < 0.01
U87MG cell gene expression profiles after FF treatment
To further investigate the molecular mechanisms underlying the antiproliferative effects of FF, we used a gene expression profile chip (BGI-Shenzhen, China) to compare changes in gene expression between control U87MG cells (0 μM for 48 h) and FF-treated U87MG cells (100 μM for 48 h). Results of our microarray study indicated that 2685 unique genes (|log2 ratio| > 2, P < 0.05), including 405 downregulated genes and 2280 upregulated genes, had significantly altered expression levels (Supplement Table). FF treatment induced the expression of proliferation- and cell cycle-associated genes such as FoxO1 (Supplement Table).
We also performed gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses to further elucidate the mechanism of the antiproliferative effects of FF using the DAVID online software. As shown in Tables 1 and 2, among the top 10 most obvious changes in GO biological process classification, three GO biological processes, namely cell cycle phase, cell cycle and M phase, were found to be related to the cell cycle. Cancer-associated pathways, such as mTOR signaling and insulin signaling pathways, were also among those with the top 10 most significant changes.
FF induces G0/G1 arrest in U87MG cells
Gene expression profiling results indicated that FF treatment (100 μM for 48 h) significantly affected molecules involved in the cell cycle. To provide evidence to support this result, we conducted cell cycle analyses using flow cytometry. Figure 2a shows the cell cycle profiles of U87MG cells after FF treatment (0, 50, or 100 μM) at 24 and 48 h. The numbers of cells that were in the G0/G1 phase increased in a time- and dose-dependent manner. These results indicated that DNA synthesis was inhibited by FF.

Cell cycle progression arrest at G0/G1 after fenofibrate treatment. a After treatment with fenofibrate at different concentrations (0, 50, and 100 μM) for 24 and 48 h, U87MG cells were used for cell cycle analyses with propidium iodide staining and flow cytometry analysis. b Fenofibrate increased PPARα transcriptional activity in a dose-dependent manner. c Western blot results for PPARα protein levels in U87MG cells treated with 100 nM siPPARα. d PPARα is involved in fenofibrate-induced (100 μM for 48 h) cell cycle arrest. **P < 0.01; *P < 0.05
Following this, we investigated whether FF induces G0/G1 arrest via a PPARα-dependent mechanism. As shown in Fig. 2b, FF significantly increased PPARα transcriptional activity in a dose-dependent manner. In addition, there was >75 % decrease in PPARα protein levels when U87MG cells were incubated with 100 nM PPARα siRNA for 48 h (Fig. 2c). Pretreating cells with PPARα siRNA duplexes significantly rescued FF-induced cell cycle arrest (Fig. 2d).
G0/G1 arrest by FF occurs in U87MG cells via FoxO1-induced p27kip upregulation
FF treatment increased the expression of FoxO1 (Supplement Table). In addition, Pearson correlation tests conducted on GBM patients’ mRNA expression data from CGGA and TCGA revealed a significant positive correlation between FoxO1 and PPARα (SFigure 1A and 1B). FoxO1 is known to bind to and activate the promoter of the cyclin-dependent kinase inhibitor p27kip [15]. Consistent with this, Western blot analyses (Figs. 3a and 4a) showed appreciable upregulation of FoxO1 and p27kip in FF-treated cells. Moreover, control cells and FF-treated cells (50 and 100 μM) exhibited significant changes in their FoxO1 nuclear contents (Fig. 3b). In addition, siPPARα treatment significantly rescued FF-induced upregulation and nuclear translocation of FoxO1 (Fig. 3c, d)

Fenofibrate effects on FoxO1 subcellular localization and expression. a Total proteins were isolated from cells after treatment with fenofibrate (0, 50, and 100 μM) for 48 h, and FoxO1 expression was assessed by Western blot analysis. b Quantification of FoxO1 nuclear localization in exponentially growing U87MG cells after treatment with fenofibrate (0, 50, or 100 μM) for 48 h. c, d Cells were treated with 100 μM fenofibrate in the presence of siPPARα for 48 h. HDAC1 expression was used as nuclear control. **P < 0.01

Evaluation of involvement of the FoxO1–p27kip axis in fenofibrate-induced G0/G1 arrest. a Western blot analysis for p27kip protein levels in U87MG cells after treatment with fenofibrate (0, 50, or 100 μM) for 48 h. b Western blot results for FoxO1 protein levels in U87MG cells treated with 100 nM siFoxO1. c Rescue experiment performed by introducing siFoxO1 into U87MG cells in the presence or absence of fenofibrate. Western blot results for p27kip expression in the indicated cells. **P < 0.01
Following this, we assessed whether FF induces cell cycle arrest via the FoxO1–p27kip axis. The effect of siFoxO1 knockdown was first tested by Western blot analysis (Fig. 4b). U87MG cells were then treated with siFoxO1 and subsequently exposed to FF (100 μM) for 48 h. The effects of FF on p27kip expression and cell cycle arrest of GBM U87MG cells were partially rescued by siFoxO1 (Fig. 4c). These results indicated that the FoxO1–p27kip axis was involved in FF-induced G0/G1 arrest in GBM U87MG cells.
Discussion
FF is a member of the fibrate family and has been shown to have potent antiproliferative effects in cancer cells. In this study, we found that FF treatment induced G0/G1 arrest in U87MG cells. There are two possible mechanisms underlying drug-induced cell cycle arrest. One is drug–DNA adduct generation, and the other is drug-induced interference of growth signals. With regard to the first possibility, it has been reported that G0/G1 arrest and G2/M arrest are induced by mechlorethamine and melphalan, both of which are alkylating agents [18, 19]. However, FF has no alkylating moiety. Moreover, FF cannot covalently bind to DNA [20]. With regard to the second possibility, some studies have shown that FF can inhibit cancer cell growth by blocking the growth signaling pathways. Urbanska et al. reported that FF inhibits IGF-I-mediated cancer cell growth and survival depending on activation of PPARα [10]. Besides IGF-I-mediated growth signaling pathways, NF-κB and PI3K/Akt signaling pathways are also involved in FF-induced apoptosis and antiproliferation [21, 22]. Recently, Wilk et al. showed that FF can cause human glioblastoma cell death through the AMPK–mTOR–autophagy pathway [23], which is consistent with the results of our KEGG analyses. Moreover, signal transduction inhibitors can induce G0/G1 arrest and G2/M arrest [24]. Consistent with this idea, FF-induced nuclear translocation of FoxO1 was found to depend on activation of PPARα in our results.
Aberrant activation of PI3K/Akt signaling is involved in human glioma cell proliferation, and its downstream effectors are promising targets for glioma cancer therapy [25]. The FoxO proteins (FoxO1, FoxO3a, FoxO4, and FoxO6) are key effectors of PI3K/Akt signaling and regulate many biological processes such as cell cycle progression and cell differentiation [16, 26]. In addition, as a transcription factor, FoxO1 acts at a convergence point of several growth factor receptor tyrosine kinase pathways that may play central roles in glioma tumorigenesis [16, 17]. In a previous study, Chen et al. found that FF-induced lipid-lowering effects were associated with increased expression of FoxO1 in myotubes [27]. Our study provides a direct link between FF treatment and FoxO1 nuclear accumulation, which in turn increases the amount of a FoxO1-dependent cell cycle-related protein, p27kip [15]. Two pathways for p27kip account for G1-to-S phase progression. The first involves inhibition of cyclin E–cdk2. In the second pathway, p27 enhances the activation and nuclear localization of cyclin D–cdk complexes during the early G1 phase [28, 29]. Thus, transactivation of p27kip could inhibit cancer cell growth [30]. It has been reported that FF treatment resulted in cell cycle arrest at the G0/G1 phase accompanied by p27kip upregulation in breast cancer cells [22]. In line with these results, we found that FF could also upregulate the expression of p27kip. In addition, siFoxO1 significantly blocked the effect of FF-induced p27kip expression and the cell cycle arrest of U87MG cells. Taken together, these results led us to conclude that the FoxO1–p27kip pathway is highly important for FF-induced anticancer effects.
In summary, we showed that FF, a PPARα agonist, could inhibit U87MG cell proliferation and induce cell cycle arrest via the PPARα/FoxO1/p27kip pathway. This pathway needs to be studied further to better understand the anticancer effects of FF.
References
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352:987–96.
Drappatz J, Norden AD. Wen PY Therapeutic strategies for inhibiting invasion in glioblastoma. Expert Rev Neurother. 2009;9:519–34.
Blane GF. Review of European clinical experience with fenofibrate. Cardiology. 1989;76 Suppl 1:1–10. discussion 10–13.
Jiao H. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol Appl Pharmacol. 2002;185:172–9.
Saidi SA, Holland CM, Charnock-Jones DS, et al. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol Cancer. 2006;5:13.
Panigrahy D, Kaipainen A, Huang S, et al. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A. 2008;105:985–90.
Drukala J, Urbanska K, Wilk A, et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARα -mediated inhibition of glioma cell motility in vitro. Mol Cancer. 2010;9:159.
Grabacka M, Placha W, Plonka PM, et al. Inhibition of melanoma metastases by fenofibrate. Arch Dermatol Res. 2004;296:54–8.
Grabacka M. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028–36.
Urbanska K, Pannizzo P, Grabacka M, et al. Activation of PPARα inhibits IGF-I‐mediated growth and survival responses in medulloblastoma cell lines. Int J Cancer. 2008;123:1015–24.
Guttilla IK. White BA Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204–16.
Goto T, Takano M, Albergaria A, et al. Mechanism and functional consequences of loss of FOXO1 expression in endometrioid endometrial cancer cells. Oncogene. 2008;27:9–19.
Dong XY, Chen CS, Sun XD, et al. FOXO1A is a candidate for the 13q14 tumor suppressor gene inhibiting androgen receptor signaling in prostate cancer. Cancer Res. 2006;66:6998–7006.
Kojima T, Shimazui T, Horie R, et al. FOXO1 and TCF7L2 genes involved in metastasis and poor prognosis in clear cell renal cell carcinoma. Gene Chromosome Canc. 2010;49:379–89.
Machida S, Spangenburg EE. Booth FW Forkhead transcription factor FOXO1 transduces insulin-like growth factor’s signal to p27Kip1 in primary skeletal muscle satellite cells. J Cell Physiol. 2003;196:523–31.
Aoki M, Jiang H. Vogt PK Proteasomal degradation of the FoXO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci U S A. 2004;101:13613–7.
Lau CJ, Koty Z. Nalbantoglu J Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009;69:5433–40.
Lupi M, Cappella P, Matera G, et al. Interpreting cell cycle effects of drugs: the case of melphalan. Cancer Chemoth Pharm. 2006;57:443–57.
Gorczyca W, Gong J, Ardelt B, et al. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res. 1993;53:3186–92.
von Daniken A, Lutz WK. Schlatter C Lack of covalent binding to rat liver DNA of the hypolipidemic drugs clofibrate and fenofibrate. Toxicol Lett. 1981;7:305–10.
Zeng R, Xiong Y, Zhu FM, et al. Fenofibrate attenuated glucose-induced mesangial cells proliferation and extracellular matrix synthesis via PI3K/AKT and ERK1/2. Plos One. 2013;8:e76836.
Li T, Zhang Q, Zhang J, et al. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-kappaB pathway. BMC Cancer. 2014;14:96.
Wilk A, Wyczechowska D, Zapata A, et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol. 2014;57:744.
Yamasaki F, Hama S, Yoshioka H, et al. Staurosporine-induced apoptosis is independent of p16 and p21 and achieved via arrest at G2/m and at G1 in U251MG human glioma cell line. Cancer Chemother Pharmacol. 2003;51:271–83.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62.
Huang HJ. Tindall DJ Dynamic FoXO transcription factors. J Cell Sci. 2007;120:2479–87.
Chen W-L, Chen Y-L, Chiang Y-M, et al. Fenofibrate lowers lipid accumulation in myotubes by modulating the PPARα/AMPK/FoXO1/ATGL pathway. Biochem Pharmacol. 2012;84:522–31.
Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60.
Ciarallo S, Subramaniam V, Hung W, et al. Altered p27(Kip1) phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor beta-mediated G(1) arrest. Mol Cell Biol. 2002;22:2993–3002.
Schiappacassi M, Lovat F, Canzonieri V, et al. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer Ther. 2008;7:1164–75.
Acknowledgments
This work was supported by grants from the National High Technology Research and Development Program of China (863) (2012AA02A508), Research Special Fund For Public Welfare Industry of Health (201402008), National Natural Science Foundation of China (91229121, 81272792, 81472362,81172389, 81372709, 81302185), Jiangsu Province’s Natural Science Foundation (20131019), Jiangsu Province’s Key Provincial Talents Program (RC2011051), Jiangsu Province’s Key Discipline of Medicine (XK201117), Jiangsu Provincial Special Program of Medical Science (BL2012028), Program for Development of Innovative Research Team in the First Affiliated Hospital of NJMU, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Dong-feng Han, Jun-xia Zhang, and Wen-jin Wei contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
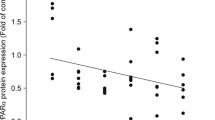
SFigure 1
Positive correlation between FoxO1 and PPARα in GBM samples. (A) FoxO1 showed positive correlation with PPARα in 89 GBM patients downloaded from CGGA. (B) FoxO1 showed positive correlation with PPARα in 169 GBM patients downloaded from TCGA. (TIFF 149 kb)
ESM 2
(XLSX 183 kb)
Rights and permissions
About this article

Cite this article
Han, Df., Zhang, Jx., Wei, Wj. et al. Fenofibrate induces G0/G1 phase arrest by modulating the PPARα/FoxO1/p27kip pathway in human glioblastoma cells. Tumor Biol. 36, 3823–3829 (2015). https://doi.org/10.1007/s13277-014-3024-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-3024-4