
Abstract
MicroRNAs are short regulatory RNAs that play crucial roles in cancer development and progression. MicroRNA-646 (miR-646) is downregulated in many human cancers, and increasing evidence indicates that it functions as a tumor suppressor. However, the role of miR-646 in osteosarcoma remains unclear. Expression levels of miR-646 in osteosarcoma cell lines and patient tissues were evaluated by quantitative real-time PCR (qRT-PCR), and the clinicopathological significance of the resultant data was later analyzed. Next, we investigated the role of miR-646 to determine its potential roles on osteosarcoma cell proliferation, migration, and invasion in vitro. A luciferase reporter assay was conducted to confirm the target gene of miR-646, and the results were validated in the osteosarcoma cell line. In this study, we found that miR-646 was downregulated in osteosarcoma cell lines and osteosarcoma tissues compared with normal osteoblast cell line NHOst and paired adjacent nontumor tissue. We found that a lower expression of miR-646 was associated with metastasis. In osteosarcoma cells, overexpression of miR-646 inhibited cell proliferation, migration, and invasion. In contrast, downregulation of miR-646 expression promoted osteosarcoma cell proliferation, migration, and invasion. Next, we identified that the FGF2 gene is a novel direct target of miR-646 in osteosarcoma cells. Moreover, enforced expression of FGF2 partially reversed the inhibition of cell proliferation, migration, and invasion that was caused by miR-646. Our study demonstrated that miR-646 might be a tumor suppressor in osteosarcoma via the regulation of FGF2, which provided a potential prognostic biomarker and therapeutic target.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Osteosarcoma is the most frequent malignant primary bone tumor, and it arises primarily in the metaphysis of the long bones in children and adolescents [1]. The 5-year survival rate of osteosarcoma patients has significantly improved over the past several decades to approximately 60–70 % since the neoadjuvant and adjuvant chemotherapy [2]. However, the outcomes remain poor for most osteosarcoma patients with metastasis or recurrence [3]. Therefore, there is an urgent need to identify the molecular mechanisms of osteosarcoma.
MicroRNAs (miRNAs) are a class of small (22-nucleotide) noncoding RNA molecules that control gene expression by binding to the 3′ untranslated region (UTR) of their target messenger RNAs (mRNAs) and play an important role in carcinogenesis [4]. Accumulating evidences implicated that miRNAs play key roles in many biological processes, including cell proliferation, apoptosis, angiogenesis, migration, and invasion [5]. Furthermore, the aberrant expression of specific miRNAs has been found to be associated with the development and clinical outcomes of various cancers [6–8]. Recent studies demonstrated that several miRNAs have been implicated in the development and progression of osteosarcoma, such as miR-33a, miR-100, and miR-221 [9–11]. However, miRNAs and their roles in osteosarcoma tumorigenesis are still largely unknown.
miR-646 was first identified in a study of miRNAs expressed in human cerebral cortical gray and white matter. Gatsiou showed that miR-646 belongs to the miR-15/107 gene group, and dysfunction in miR-107 expression may contribute to neoplasia, neurodegeneration, cardiovascular dysfunction, and other diseases [12]. Wang found that miR-646 was expressed in a higher level in the serum of cervical squamous cell carcinoma patients after surgery than that before surgery, which indicated that the circulating miR-646 could potentially serve as noninvasive biomarkers [13]. Li’s study showed that miR-646 was downregulated in renal cancer and low expression of miR-646 was significantly associated with distant metastasis. Furthermore, downregulated miR-646 in renal cancer was associated with tumor metastasis through the MAPK pathway by targeting NOB1 [14]. However, the dysregulation of miR-646 and its possible involvement in osteosarcoma have not been reported.
Materials and methods
Patients and specimens
A total of 64 osteosarcoma tissues and matched nontumor bone tissues (located 3 cm away from the tumor) were obtained from patients who underwent surgery in the Department of Orthopedics, The First Affiliated Hospital of Xinxiang Medical University, between 2006 and 2010. None of the patients received radiotherapy or chemotherapy before surgery. After surgical resection, tumor specimens and paired adjacent nontumor specimens were collected and immediately stored in liquid nitrogen until use. All osteosarcoma specimens and paired adjacent nontumor specimens were confirmed by a senior pathologist. For the use of these clinical materials for research purposes, prior patient’s consent and approval from the Institute Research Ethics Committee were obtained.
Cell culture and cell transfection
Normal osteoblast cell line NHOst was purchased from the American Type Culture Collection (ATCC, USA). Human osteosarcoma cell lines MG-63, SAOS-2, HOS, and U2OS were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (CCCAS, China). NHOst cells were cultured in Ham’s F12/DMEM (Gibco), and other cells were cultured in RPMI-1640 (Gibco) with 10 % fetal bovine serum (FBS, Gibco), 50 U/ml of penicillin, and 50 μg/ml of streptomycin. All cells were cultured in a sterile incubator maintained at 37 °C with 5 % CO2.
MG-63 cells were seeded in 12-well plates and incubated overnight, then transiently transfected with miR-646 mimic, scramble mimic, miR-646 inhibitor, and miR-negative control of inhibitor (anti-miR-Ctrl) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. miR-646 mimic and miR-646 inhibitor were obtained from Applied Biosystems. The full-length FGF2 cDNA (which included the ORF and 3′ UTR) was PCR amplified and cloned into the pcDNA3.1 vector to generate the pcDNA-FGF2 constructs that were used in the rescue assays. MG-63 cells in 12-well plates were cotransfected with miR-646 mimic and the pcDNA-FGF2 plasmid DNA.
Cell proliferation assay
The in vitro cell proliferation of osteosarcoma cells was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method. In brief, cells were seeded into 96-well plates and transfected with miR-646 mimic or miR-646 inhibitor for 48 h. In the indicated time periods, 0.1 ml of spent medium was replaced with an equal volume of fresh medium containing MTT 0.5 mg/ml. Plates were incubated at 37 °C for 4 h, and then, the medium was replaced with 0.1 ml of DMSO (Sigma), and plates were agitated at room temperature for 10 min. The absorbance was measured at 490 nm using an enzyme-labeled analyzer. Three independent experiments (three replicates in each) were performed.
Wound healing assay
To determine cell migration, osteosarcoma cells transfected with miR-646 mimic or miR-646 inhibitor were seeded into 12-well plates and allowed to grow to 90–95 % confluence. Before scratching, cells were starved for 24 h in the medium with 1 % FBS. Similar-sized wounds were introduced to monolayer cells using a sterile white pipette tip. Wounded monolayer cells were washed three times by PBS to remove cell debris and then cultured. The speed of wound closure was monitored and photographed at 48 h.
Transwell invasion assay
To determine cell invasion, Matrigel-coated invasion chambers (8 μm, Costar) were used according to the manufacturer’s protocol. Briefly, miR-646 mimic or miR-646 inhibitor transfected cells were harvested, resuspended (1 × 105 cells/well) in 200-μl serum-free medium, and transferred to the upper chamber of the Matrigel-coated inserts; culture medium containing 10 % FBS was placed in the bottom chamber. The cells were incubated for 24 h at 37 °C. After incubation, the noninvaded cells on the upper membrane surface were removed with a cotton tip, and the cells that passed through the filter were fixed and stained using 0.1 % crystal violet. Numbers of invaded cells were counted in five randomly selected fields under a microscope (Nikon).
Prediction of miRNAs targeting FGF2
miRNA target predicting algorithms miRDB (http://mirdb.org/miRDB/), TargetScan (http://www.targetscan.org/), and PicTar (http://pictar.mdc-berlin.de/) were used to predict miRNAs targeting FGF2 and the binding regions.
Luciferase reporter assay
The 3′ untranslated region (UTR) of the human FGF2 gene that was predicted to interact with miR-646 was synthesized and inserted into pMIR-REPORT (Ambion), yielding pMIR-REPORT FGF2. Mutations within potential miR-646 binding sites were generated by nucleotide replacement of wild-type sequence to inhibit miR-646 binding. Cells were transfected with the pMIR-REPORT vectors containing the 3′ UTR variants and miR-646 mimics for 24 h. Luciferase values were determined using the Dual-Luciferase Reporter Assay System (Promega).
RNA isolation and quantitative real-time PCR
Total RNA from tissue samples and cell lines were harvested using the miRNA Isolation Kit (Ambion). Expression of mature miRNAs was assayed using Taqman MicroRNA Assay (Applied Biosystems) specific for hsa-miR-646. Briefly, 10 ng of total RNA was reverse transcribed to cDNA with specific stem-loop RT primers. Quantitative real-time PCR was performed by using an Applied Biosystems 7900 Real-time PCR system and a TaqMan Universal PCR Master Mix. All the primers were obtained from the TaqMan miRNA Assays. Small nuclear U6 snRNA (Applied Biosystems) was used as an internal control. All reactions were performed at least in triplicate.
Western blot analysis
Western blot analysis was carried out using standard methods. Proteins were separated by 10 % SDS-PAGE and then transferred to PVDF membranes (Amersham). Membranes were blocked overnight with 5 % nonfat dried milk and incubated for 2 h with anti-FGF2 antibody (Abcam) at a 1:1000 dilution or with anti-β-actin antibody (Abcam) at a 1:10,000 dilution. After washing with TBST, the membranes were incubated for 2 h with a goat anti-rabbit antibody (Abcam) at either a 1:5000 dilution or a 1:50,000 dilution.
Statistical analysis
All statistical analyses were performed using SPSS version 18.0 software (IBM). Data are expressed as the mean ± SD from at least three separate experiments. Data were analyzed using Student’s t test or one-way ANOVA. Values of P < 0.05 were considered statistically significant.
Results
Downregulation of miR-646 in osteosarcoma cell lines and tissues
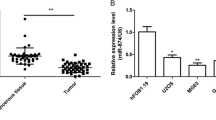
First, we investigated the expression of miR-646 in four osteosarcoma cell lines (MG-63, SAOS-2, HOS, U2OS) and the human osteoblastic cell line NHOst by qRT-PCR. Our results indicated that the osteosarcoma cell lines exhibited a significantly lower expression of miR-646 compared to osteoblastic cell line NHOst (P < 0.05, Fig. 1a). Furthermore, we examined the expression of miR-646 in 64 pairs of human osteosarcoma tissues and matched nontumor tissues. Our data showed that the expression level of miR-646 was significantly downregulated in osteosarcoma tissues in comparison to the adjacent nontumor tissues (P < 0.05, Fig. 1b). We then determined whether miR-646 was associated with tumor metastasis. Tumor samples were divided into two groups (metastases and nonmetastases) according to their metastatic status. We found that the expression of miR-646 in the metastatic osteosarcoma tissues was significantly lower than that in the nonmetastatic tissues (P < 0.05, Fig. 1c). Taken together, these results supported the notion that miR-646 may act as a tumor suppressor in osteosarcoma and play a key role in osteosarcoma metastasis.

The expression of miR-646 in human osteosarcoma cell lines and tissues. a The expression of miR-646 in osteosarcoma cell lines (MG-63, SAOS-2, HOS, and U2OS) were lower than that in normal osteoblast cell line NHOst. b The expression of miR-646 in the osteosarcoma tissues was lower than that in the adjacent nontumor tissues. c The expression of miR-646 in the metastatic osteosarcoma tissues was lower than that in the nonmetastases tissues. Data are presented as mean ± SD. *P < 0.05
miR-646 suppresses osteosarcoma cell proliferation, migration, and invasion
To explore the role of miR-646 in the development of osteosarcoma, we transfected with miR-646 mimic or miR-646 inhibitor into MG63 cells; expression of miR-646 was confirmed by qRT-PCR (P < 0.05, Fig. 2a, b). MTT assay showed that the proliferation rate of MG-63 cells transfected with miR-646 mimic was significantly decreased in comparison to cells transfected with scramble mimic (P < 0.05, Fig. 2c). In contrast, the proliferation rate of MG63 cells transfected with miR-646 inhibitor was significantly increased in comparison to cells transfected with anti-miR-Ctrl (Fig. 2d, P < 0.05). Furthermore, to analyze the role of miR-646 in cell migration and invasion, wound healing assay and transwell invasion assay were performed with MG-63 cells. Wound healing assay showed that miR-646 mimic markedly inhibited osteosarcoma cell migration (Fig. 2e, P < 0.05). Consistent with this result, silencing of miR-646 resulted in a significant increase in osteosarcoma cell migration (P < 0.05, Fig. 2f). Transwell invasion assays indicated that the invasion potential of osteosarcoma cells transfected with miR-646 mimics was significantly decreased (Fig. 2g, P < 0.05), whereas silencing of miR-646 significantly enhanced osteosarcoma cell invasion (Fig. 2h, P < 0.05). Taken together, these data demonstrated that miR-646 acting as a tumor suppressor could inhibit osteosarcoma cell proliferation, migration, and invasion.

miR-646 regulates osteosarcoma cell proliferation, migration, and invasion. a Upregulation of miR-646 by transfection with miR-646 mimic in MG-63 cells. b Downregulation of miR-646 by transfection with miR-646 inhibitor in MG-63 cells. c, d MTT assay was performed to analyze the effect of miR-646 on cell proliferation of MG-63 cells. e, f Wound healing assay was used to analyze the effect of miR-646 on cell migration of MG-63 cells. g, h Transwell invasion assay was utilized to analyze the effect of miR-646 on cell invasion of MG-63 cells. Data are presented as means ± SD from three independent experiments. *P < 0.05
miR-646 directly targets the FGF2 in osteosarcoma cells
To investigate the mechanism of miR-646 function in osteosarcoma carcinogenesis, we used a bioinformatic analysis to search for putative protein-coding gene targets of miR-646; according to this rationale, FGF2 was selected as one of the candidate targets of miR-646. Next, we performed a luciferase reporter assay to determine whether FGF2 was a direct target of miR-646 in osteosarcoma cells. The target region sequence of FGF2 3′ UTR (Wt 3′ UTR) or the mutant sequence (Mut 3′ UTR) was cloned into a luciferase reporter vector (Fig. 3a). These constructed reporter vectors were cotransfected with miR-646 mimic or scramble mimic into the MG-63 cell line. Our findings indicated that miR-646 could downregulate the luciferase activity of the FGF2 Wt 3′ UTR construct (Fig. 3b, P < 0.05). The luciferase reporter data showed that FGF2 is a specific target of miR-646.

miR-646 negatively regulates FGF2 expression in osteosarcoma cells. a A schematic representation of the FGF2 3′ UTR that showed the putative miRNA target site. b Luciferase assay. MG-63 cells were transiently cotransfected with Wt/Mut 3′ UTR with miRNAs as indicated. c Overexpression of miR-646 inhibited the expression of FGF2 at the mRNA and protein level in MG-63 cells. d Knockdown miR-646 expression increased the expression of FGF2 at the mRNA and protein level in MG-63 cells. Data are presented as means ± SD from three independent experiments. *P < 0.05
To further confirm that FGF2 is a target gene for miR-646, qRT-PCR and Western blot analysis were used to detect the expression of FGF2 regulated by miR-646 in MG-63 cells. Our data revealed that FGF2 mRNA and protein expression was significantly downregulated after enforced expression of miR-646 in MG-63 cells (Fig. 3c, P < 0.05). In contrast, FGF2 mRNA and protein expression was upregulated after the expression of miR-646 in MG-63 cells was knocked down (Fig. 3d, P < 0.05).
Overexpression of FGF2 impairs the miR-646-induced suppression of osteosarcoma cell proliferation, migration, and invasion
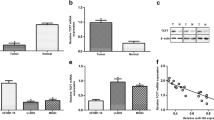
To assess the regulation of miR-646 on FGF2 expression, the expression level of FGF2 was analyzed in osteosarcoma tissues and cell lines. Our results showed that the expression level of FGF2 was upregulated in osteosarcoma tissues compared to adjacent normal tissues (P < 0.05, Fig. 4a, b). In addition, miR-646 was significantly increased in osteosarcoma cell lines compared with the osteoblast NHOst cell line (P < 0.05, Fig. 4c, d). We then performed rescue experiments to further validate that the targeting of FGF2 was involved in the antitumor properties of miR-646 in osteosarcoma cell line MG-63. The FGF2 expression vector pcDNA-FGF2 was used to restore FGF2 expression. Overexpression of FGF2 could reverse the effect of miR-646 on the inhibition of proliferation, migration, and invasion (P < 0.05, Fig. 4e–g). Taken together, these findings indicated that FGF2 is a functionally important target of miR-646 that is involved in the proliferation, migration, and invasion of osteosarcoma cells.

FGF2 is required for miR-646-directed osteosarcoma cell proliferation, migration, and invasion. a, b FGF2 expression was increased in the osteosarcoma tissues compared to the adjacent nontumor tissues. c, d FGF2 expression was increased in the osteosarcoma cell lines compared to osteoblast cell line NHOst. e MTT assay was used to detect in MG-63 cells cotransfected with miR-646 mimic and pcDNA-FGF2 plasmid. f Wound healing assay was performed to detect the effects on cell migration of MG-63 cells treated as described in e, g. Transwell invasion assay was utilized to detect the effects on cell invasion of MG-63 cells treated as described in e. Data are presented as means ± SD from three independent experiments. *P < 0.05
Discussion
Most cancer deaths are caused by complications that arise from metastasis. The targeting of metastatic disease is therefore a pivotal anticancer strategy [15]. Increasing evidences indicated that miRNAs may play key roles in the tumorigenic processes such as tumor invasion and metastasis through the regulation of a variety of genes [16–18]. In the present study, we investigated the roles of miR-646 in osteosarcoma cell growth and metastasis. Our results indicated that miR-646 expression was significantly downregulated in osteosarcoma cell lines and tissue comparison with osteoblast cell line NHOst and adjacent nontumor tissues. Statistical analyses showed that the expression level of miR-646 was correlated with osteosarcoma metastasis. Further studies indicated that overexpression of miR-646 was able to inhibit cell proliferation, migration, and invasion in MG-63 cells. In contrast, downregulation of miR-646 could promote cell proliferation, migration, and invasion in MG-63 cells. Therefore, these data for the first time suggested that miR-646 could act as a tumor suppressor in the progression of osteosarcoma. However, further studies are still needed to investigate its underlying mechanism.
Fibroblast growth factor 2 (FGF2) is well characterized, bearing all typical features of the fibroblast growth factor family, and is regarded as a prototypic growth factor [19]. FGF2 is upregulated in lots of human carcinomas and plays important roles in carcinogenesis, such as cancer cell proliferation and metastasis [20]. Kuhn showed that FGF2 expression levels were significantly higher in lung cancer patients and were correlated to tumor progression and poor prognosis [21]. Recent studies showed that FGF2 was significantly increased in prostate cancer and played an important role in prostate carcinogenesis and malignant progression, and knockout of FGF2 was found to delay tumor progression in transgenic adenocarcinoma of the mouse prostate model [22, 23]. Li found that FGF-Trap effectively suppressed FGF-2-induced proliferation and migration of human umbilical vein endothelial cells, and FGF-Trap potently inhibited tumor growth and angiogenesis in Caki-1 and A549 xenograft models in vivo [24]. Zhou found that miR-503 targeted FGF2 and VEGFA and inhibited tumor angiogenesis and growth in hepatocellular carcinomas [25]. Ren’s study found that apurinic/apyrimidinic endonuclease 1 induced upregulation of FGF2 and its receptor 3 induces angiogenesis in human osteosarcoma cells [26]. However, the underlying mechanisms are still unclear. In our study, we indicated that FGF2 is a direct target of miR-646 and found that miR-646 overexpression is correlated with FGF2 downregulation leading to the inhibition of cell proliferation, migration, and invasion in osteosarcoma cells. These findings suggested that the tumor suppressor role of miR-646 is mediated by the regulation of FGF2 expression.
Taken together, our study indicated novel evidence that miR-646 is significantly decreased in tumor tissues and appears to function as a tumor suppressor in osteosarcoma through the regulation of FGF2 expression. Understanding the miRNA-mediated tumor suppressor pathways in human osteosarcoma may provide new information on potential therapeutic targets in the treatment of osteosarcoma.
References
Ottaviani G, Jaffe N. The epidemiology of osteosarcoma[M]//pediatric and adolescent osteosarcoma. US: Springer; 2010. p. 3–13.
Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115(7):1531–43.
Osaki M, Takeshita F, Sugimoto Y, et al. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol Ther. 2011;19(6):1123–30.
Yates LA, Norbury CJ, Gilbert RJC. The long and short of microRNA. Cell. 2013;153(3):516–9.
Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27(34):5848–56.
Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–58.
Chow TF, Mankaruos M, Scorilas A, et al. The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol. 2010;183(2):743–51.
Farazi TA, Hoell JI, Morozov P, et al. MicroRNAs in human cancer[M]//microRNA cancer regulation. Netherlands: Springer; 2013. p. 1–20.
Zhou Y, Huang Z, Wu S, et al. miR-33a is up-regulated in chemoresistant osteosarcoma and promotes osteosarcoma cell resistance to cisplatin by down-regulating TWIST. J Exp Clin Cancer Res: CR. 2014;33(1):12.
Huang J, Gao K, Lin J, et al. MicroRNA-100 inhibits osteosarcoma cell proliferation by targeting Cyr61. Tumor Biol. 2014;35(2):1095–100.
Zhao G, Cai C, Yang T, et al. MicroRNA-221 induces cell survival and cisplatin resistance through PI3K/Akt pathway in human osteosarcoma. PLoS One. 2013;8(1):e53906.
Finnerty JR, Wang WX, Hébert SS, et al. The miR-15/107 group of microRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J Mol Biol. 2010;402(3):491–509.
Wang WT, Zhao YN, Yan JX, et al. Differentially expressed microRNAs in the serum of cervical squamous cell carcinoma patients before and after surgery. J Hematol Oncol. 2014;7(1):6.
Li W, Liu M, Feng Y, et al. Downregulated miR-646 in clear cell renal carcinoma correlated with tumour metastasis by targeting the nin one binding protein (NOB1). Br J Cancer, 2014.
Zhang H, Zhang Y, Duan HO, et al. TIP30 is associated with progression and metastasis of prostate cancer. Int J Cancer. 2008;123(4):810–6.
Paterson EL, Kazenwadel J, Bert AG, et al. Down-regulation of the miRNA-200 family at the invasive front of colorectal cancers with degraded basement membrane indicates EMT is involved in cancer progression. Neoplasia. 2013;15(2):180–IN22.
Qi L, Bart J, Tan LP, et al. Expression of miR-21 and its targets (PTEN, PDCD4, TM1) in flat epithelial atypia of the breast in relation to ductal carcinoma in situ and invasive carcinoma. BMC Cancer. 2009;9(1):163.
Ma L, Young J, Prabhala H, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–56.
Cronauer MV, Schulz WA, Seifert HH, et al. Fibroblast growth factors and their receptors in urological cancers: basic research and clinical implications. Eur Urol. 2003;43(3):309–19.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29.
Kuhn H, Köpff C, Konrad J, et al. Influence of basic fibroblast growth factor on the proliferation of non-small cell lung cancer cell lines. Lung Cancer. 2004;44(2):167–74.
Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5(5):1063–71.
Polnaszek N, Kwabi-Addo B, Peterson LE, et al. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 2003;63(18):5754–60.
Li D, Wei X, Xie K, et al. A novel decoy receptor fusion protein for FGF-2 potently inhibits tumour growth. Br J Cancer, 2014.
Zhou B, Ma R, Si W, et al. MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett. 2013;333(2):159–69.
Ren T, Qing Y, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 induced upregulation of fibroblast growth factor 2 and its receptor 3 induces angiogenesis in human osteosarcoma cells. Cancer Sci. 2014;105(2):186–94.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sun, Xh., Geng, Xl., Zhang, J. et al. miRNA-646 suppresses osteosarcoma cell metastasis by downregulating fibroblast growth factor 2 (FGF2). Tumor Biol. 36, 2127–2134 (2015). https://doi.org/10.1007/s13277-014-2822-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2822-z