
Abstract
Histone deacetylases (HDACs) play a critical role in the proliferation, differentiation, and apoptosis of cancer cells. An obstacle for the application of HDAC inhibitors as effective anti-cancer therapeutics is that our current knowledge on the contributions of different HDACs in various cancer types remains scarce. The present study reported that the mRNA and protein levels of HDAC5 were up-regulated in human hepatocellular carcinoma (HCC) tissues and cells as shown by quantitative real-time PCR and Western blot. MTT assay and BrdU incorporation assay showed that the down-regulation of HDAC5 inhibited cell proliferation in HepG2, Hep3B, and Huh7 cell lines. Data from in vivo xenograft tumorigenesis model also demonstrated the anti-proliferative effect of HDAC5 depletion on tumor cell growth. Furthermore, the suppression of HDAC5 promoted cell apoptosis and induced G1-phase cell cycle arrest in HCC cells. On the molecular level, we observed altered expression of apoptosis-related proteins such as p53, bax, bcl-2, cyto C, and caspase 3 in HDAC5-shRNA-transfected cells. Knockdown of HDAC5 led to a significant up-regulation of p21 and down-regulation of cyclin D1 and CDK2/4/6. We also found that the down-regulation of HDAC5 substantially increased p53 stability and promoted its nuclear localization and transcriptional activity. Our study suggested that knockdown of HDAC5 could inhibit cancer cell proliferation by the induction of cell cycle arrest and apoptosis; thus, suppression of HDAC5 may be a viable option for treating HCC patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is one of the most malignant tumors in the world, causing more than 600,000 deaths per year [1]. Current treatments for HCC include chemotherapy, radiotherapy, and surgical operation [2, 3]. However, tumor recurrence and metastases often occurred, and the long-term disease-free survival remains unsatisfactory even after the above treatments, indicating the necessity of developing new therapeutic strategies for HCC therapy [4]. Up to now, many efforts have been made to understand the molecular mechanisms that underlie the cellular malignancy phenotype and that are involved in the process of cancer invasion and metastasis. Investigations showed that the biological processes such as apoptosis and cell cycle arrest played a critical role in the cancer recurrence [5, 6]. However, the mechanisms underlying the disruption of these critical pathways in the tumorigenesis of HCC are still not fully elucidated.
The histone deacetylases (HDACs) are enzymes that modulate the acetylation level of histones and non-histone proteins to regulate gene expression and chromatin structure. HDAC family contains 18 human HDAC members, which are classified into classes I–IV based on their homology and structure [7, 8]. Pre-clinical investigations by targeted knockdown of individual HDAC members demonstrated the roles of class IIa HDACs (including HDAC4, 5, 7) in tumorigenesis. Several groups have shown that the down-regulation of HDAC4 suppressed cancer cell proliferation in vitro and arrested tumor growth in vivo through epigenetic modulation of p21 expression [9, 10]. Another study demonstrated that HDAC7 was a critical player in the proliferation of cancer cells [11]. Together, these findings suggest that the inhibition of class IIa HDACs might play an important role for cancer treatment. However, the effect of HDAC5 on tumor progression is largely ignored and needs to be further investigated to determine the potential therapeutic roles of class IIa HDAC members in cancer therapy.
The human HDAC5 gene, located on chromosome 17q21, has been shown to play critical roles in cell proliferation, cell cycle progression, and apoptosis in different cancers [12, 13]. The expression of HDAC5 is decreased in lung cancer tissues, making it a potential indicator of poor clinical outcome in patients with lung cancer [14]. In contrary, another group observed the up-regulation of HDAC5 in patients with high-risk medulloblastoma, which was associated with poor survival [15]. Also, HDAC5 was found aberrantly expressed in patients with liver cancer, suggesting that the dysfunction of HDAC5 might play a significant role in hepatocarcinogenesis [16].
Here, in the present study, we investigated the expression of HDAC5 in HCC patients and cell lines and further explored the possible effect of HDAC5 on cancer cell proliferation, cell cycle arrest, and apoptosis through in vitro and in vivo functional assays. Our data showed the overexpression of HDAC5 in HCC tissues and cells. Furthermore, we found that the suppression of HDAC5 by shRNA inhibited cancer cell proliferation through the induction of cell cycle arrest and apoptosis. These findings indicated that class IIa HDAC5 might be a novel therapeutic target in the HCC treatment.
Material and methods
Tissue sample
This study was approved by the Ethics Committee of the Second Affiliated Hospital, School of Medicine, Zhejiang University. All patients provided their written informed consent to participate in this study according to the Declaration of Helsinki. Twenty pairs of HCC tissues and their adjacent non-HCC tissues were obtained from patients who underwent surgical tumor resections at the Second Affiliated Hospital, School of Medicine, Zhejiang University in China from January 2012 to December 2013. Patients included 16 men and 4 women (mean age 65 years, range 43–81). None of the patients received chemotherapy, radiation, and/or alcoholization therapy before surgery. These patients were diagnosed based on clinical symptoms, serological tests, computed tomography, and pathological evaluations according to the “Primary Liver Cancer Clinical Diagnosis and Staging Criteria” [17].
Cell culture
Human HCC cell lines HepG2, Hep3B, and Huh7 and normal liver cell lines LO2 were obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM supplemented with 10 % fetal bovine serum, streptomycin (100 mg/mL), and penicillin (100 U/mL). Cultured cells were maintained at 37 °C and 5 % CO2 in a humid environment and passaged when the confluency reached 80 %.
shRNA knockdown of HDAC5
Plasmid transfection was performed using Lipofectamine (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were changed to medium without antibiotics 12 h before transfection. Transfections were carried out when cells reached 95 % confluence using a 1:3 ratio of DNA (μg)/lipofectamine (μl). HDAC5 shRNA and control shRNA lentivirus clones were purchased from Santa Cruz Biotechnology and used to infect HCC cells following selection with puromycin (2 μg/ml).
Quantitative real-time PCR
Total RNAs were isolated from cells by TRIzol reagent, and reverse transcriptions were performed by TaKaRa RNA PCR Kit (Takara, Japan) following the manufacturer’s instructions. In order to quantify the transcripts of the interest genes, quantitative real-time PCR (qRT-PCR) was performed using a SYBR Green Premix Ex Taq (TaKaRa, Tokyo, Japan) on an ABI 7500 system (Applied Biosystems, Foster, CA, USA).
MTT assay
Proliferation of cells was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. To be brief, cell density of 3 × 104 (cells/well) was seeded into 96-well plates and left to adhere overnight. HCC cells were transfected with HDAC5-shRNA for 72 h. Then 10 ml of 5 mg/ml MTT was added and incubated in the dark at 37 °C for 2 h. The absorbance was determined with the wavelength of 492 nm.
BrdU incorporation analysis
To analyze cell proliferation, BrdU incorporation was determined by using a commercially available kit (Roche, Indianapolis, IN). HCC cells with or without shRNA transfection were incubated BrdU for 6 h. Then the labeled cells were rinsed thoroughly and incubated with a fluorescein isothiocyanate (FITC)-labeled antibody against BrdU for 30 min. The stained cells were harvested and analyzed by a fluorescence microplate reader (Millipore, USA). Fold changes were normalized by the mean fluorescence intensity of the control group.
Cell cycle analysis
Cells were seeded at the density of 1.0 × 106 cells, and cell cycle distribution was analyzed by flow cytometry after transfection with shRNA for 72 h. After washing twice with PBS, cells were harvested and collected by centrifugation, followed by fixation in ice-cold 70 % ethanol at −20 °C overnight. Then, cells were collected and stained with 100-μl PI staining solution for 30 min in the dark followed by cell cycle analysis.
Apoptosis assay
Three days after transfection with HDAC5-shRNA or control shRNA, HCC cells were washed with PBS, detached with trypsin, and harvested. Apoptosis cells were detected with annexin V-FITC/PI according to the protocol of Annexin V-FITC cell Apoptosis Detection Kit (BD, USA).
Luciferase reporter assay
The pGL3-p53 firefly luciferase reporter plasmid and the internal control pRL-SV40 (Renilla luciferase) plasmid were purchased from Promega, USA. HepG2 cells were cultured in 24-well plates and co-transfected with the indicated plasmids using Lipofectamine 2000 (Invitrogen, USA). Cell lysates were analyzed for firefly activity, which was normalized to the value of pRL-SV40 activity.
Whole cell protein extraction and nuclear/cytoplasmic extraction
Whole cell lysate was centrifuged at 10,000 g for 30 min, and the resultant supernatant was harvested as whole cell extracts. Nuclear extraction and cytoplasmic extraction were prepared by using CelLytic NuCLEAR Extraction Kit (Sigma, USA) following the manufacturer’s instruction.
Western blot analysis
Cells were harvested by trypsinization, lysed in buffer, and prepared for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). After immunoblotting, the membranes were blocked in PBS/0.1 % Tween-20 with 5 % non-fat dry milk, and primary antibodies were incubated in PBS/0.1 % Tween-20 with 0.1–5 % non-fat dry milk. Antibodies directed against p53 (1:2,000), bcl-2 (1:2,000), bax (1:2,000), cyto C (1:1,000), caspases 3 (1:1,000), p21 (1:1,000), cyclin D1 (1:1,000), CDK2 (1:1,000), CDK4 (1:1,000), and CDK6 (1:1,000) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) with GAPDH (1:2,000), beta-actin (1:2,000), or lamin B (1:1,000) used as loading controls.
Measurement of p53 half-life
For p53 protein stability experiments, HepG2 cells were transfected with HDAC5-shRNA and control shRNA for 72 h; 20 μg/ml of cycloheximide (CHX) was added to inhibit protein synthesis for indicated time points. Cells were then processed as previously described for Western blot.
In vivo tumorigenicity model
Cells (2 × 106) were suspended in 150 μl of saline and subcutaneously injected into each nude mouse. The tumor size and body weight were measured for every 7 days. After 6 weeks, the mice were sacrificed and the tumors were harvested for further analysis. Six mice were recruited for each of the experimental group. Volume of the tumor was calculated as follows: tumor volume (cm3) = (a × b 2)/2, with a as the larger diameter and b as the smaller diameter [10].
Statistical analysis
Each experiment was performed in triplicate and repeated at least three times. All data were presented as mean ± SD and treated for statistics analysis by SPSS program. Comparison between groups were determined by ANOVA, and statistical significance was displayed as * (P < 0.05) or ** (P < 0.01).
Results
Up-regulation of HDAC5 in human HCC
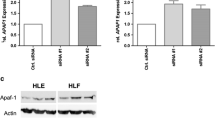
Initial studies were done to examine the relative levels of HDAC5 in 20 pairs of HCC tissues and their adjacent non-HCC tissues. As shown in Fig. 1a, the mRNA expression of HDAC5 in HCC tissues was significantly higher than that in the adjacent tissues (P < 0.05). We employed Western blot with anti-HDAC5 antibody to detect the protein expression level of HDAC5 in those clinic samples. Results also indicated that HDAC5 expression significantly increased by threefolds compared with the control (Fig. 1b, c). Then, three human HCC cell lines including HepG2, Hep3B, and Huh7 and normal liver cell lines LO2 were used to determine HDAC5 expression in vitro. qRT-PCR showed that the mRNA expression of HDAC5 in all three cell lines were significantly increased in HCC cell lines compared with normal liver cells (Fig. 1d). Western blot analysis also indicated that the HDAC5 protein expression was obviously enhanced in HCC cell lines compared to the normal liver cells (Fig. 1e, f). Collectively, these data showed that HDAC5 expression was up-regulated in HCC samples and cell lines, implying its involvement in the pathogenesis of HCC.

Up-regulation of HDAC5 in HCC tissues and cells. Representative results of qRT-PCR (a) and Western blot (b and c) analysis of HDAC5 expression in 20 pairs of HCC and adjacent non-cancerous liver tissues. Determination of mRNA and protein levels of HDAC5 by qRT-PCR (d) and Western blot (e and f) in human HCC cells and normal cells. *P < 0.05, **P < 0.01
Down-regulation of HDAC5 inhibited growth of HCC cells in vitro and in vivo
To determine whether HDAC5 could be an effective therapeutic target for HCC, the effect of HDAC5-shRNA on cell proliferation was examined. HDAC5-shRNA was transfected into HCC cell lines with lipofectamine, and HDAC5 protein expression was determined by Western blotting. Seventy-two hours after transfection, HDAC5 protein level was decreased more than half compared with control shRNA-transfected cells including HepG2 (Fig. 2a), Hep3B (Fig. 2b), and Huh7 (Fig. 2c). MTT assay demonstrated that the down-regulation of HDAC5 inhibited the proliferation of HCC cells (Fig. 2d–f). BrdU incorporation is a classic method to measure DNA synthesis and considered to be a surrogate procedure to evaluate proliferation. Not surprisingly, HDAC5-shRNA-transfected cells showed a slower growth rate consistent with the data from MTT assay (Fig. 2g–i).

Down-regulation of HDAC5 inhibited HCC cell proliferation in vitro. HCC cells were transfected with short hairpin RNA (shRNA) targeting HDAC5 for 72 h. Then, Western blot was used to analyze the expression of HDAC5 in HepG2 (a), Hep3B (b), and Huh7 (c) cells. Cell proliferation was measured using the MTT assay in HepG2 (d), Hep3B (e), and Huh7 (f) cells. And BrdU incorporation assay was also performed to determine DNA synthesis in HepG2 (d), Hep3B (e), and Huh7 (f) cells. *P < 0.05, **P < 0.01
Then, HepG2-HDAC5-shRNA cells were subcutaneously injected into nude mice, and the tumorigenicity of HDAC5 was examined. We found that both the weight and tumor volume in HepG2-HDAC5-shRNA groups were profoundly decreased compared with control groups (Fig. 3a–c), indicating that the down-regulation of HDAC5 could suppress HCC growth in vivo. Western blot analysis of subcutaneous xenografts showed that the level of HDAC5 protein in HepG2-HDAC5-shRNA xenograft was also lower than in HepG2-control xenograft (Fig. 3d). Taken together, our results demonstrated that the suppression of HDAC5 could inhibit the proliferation of hepatocellular carcinoma both in vitro and in vivo.

Down-regulation of HDAC5 inhibited HCC cell proliferation in vivo. a Representative images of resected tumors in HepG2-control and HepG2-shHDAC5 groups at the end of the experiment. Tumor weight (b) and size (c) of subcutaneous xenografts were measured with 7-day interval between HepG2-control and HepG2-shHDAC5 groups. d HDAC5 protein levels were determined between the subcutaneous tumor xenografts from HepG2-control and HepG2-shHDAC5 groups. *P < 0.05, **P < 0.01
Effect of HDAC5 suppression on apoptosis and cell cycle
As observed above, the down-regulation of HDAC5 can inhibit HCC cell growth in vitro and in vivo. We then explored the mechanisms underlying the growth inhibition by HDAC5. Because cell proliferation is closely associated to apoptosis and cell cycle progression, we analyzed the apoptosis and cell cycle in HCC cells transfected with HDAC5-shRNA. Flow cytometry analysis showed that the down-regulation of HDAC5 induced apoptosis in all three HCC cell lines including HepG2 (Fig. 4a), Hep3B (Fig. 4b), and Huh7 (Fig. 4c). These data suggested that the growth inhibitory activity of HDAC5 down-regulation was partly attributed to an increase in cell death. In addition, the percentage of the G1 phase of shRNA-HDAC5-transfected cells was consistently higher than that of controls. Meanwhile, the number of cells in S phase decreased compared with the levels in the controls (Fig. 4d), suggesting that HDAC5 inhibition induced G1 cell cycle arrest in HCC cells.

Effect of HDAC5 inhibition on apoptosis and cell cycle distribution. Three HCC cell lines were transfected with shRNA targeting HDAC5 for 72 h. a–c Results are expressed as the percentage of the number of apoptosis cells compared with the total number of cells. d Flow cytometry was used to analyze the cell cycle distribution (G1 phase: left; S phase: right) of HepG2-control and HepG2-shHDAC5 cells. *P < 0.05, **P < 0.01
Down-regulation of HDAC5 mediated protein expression related to cell cycle arrest and apoptosis
To understand the molecular basis for HDAC5 decrease-induced cell cycle arrest and apoptosis in the cells, we assayed the expression levels of apoptosis and cell cycle-related proteins with Western blot. As shown in Fig. 5a, the expression of tumor suppressor gene p53 was significantly increased in HepG2-shHDAC5 cells at 48 and 72 h post-transfection. Moreover, HDAC5 suppression resulted in the increased expression of bax, cyto C, and caspase 3 and decreased expression of bcl-2 (Fig. 5a). Besides, we found that the down-regulation of HDAC5 increased the expression of p21 and decreased the expression of cyclin D1 and CDK2/4/6 (Fig. 5b). Taken together, these results demonstrated that the inhibition of HDAC5 modulated the expression of and pro/anti-apoptotic signals and critical cell cycle regulators, leading to apoptosis and cell cycle arrest.

Effect of HDAC5 depletion on the expression of apoptosis- and cell cycle-related protein. Cell lysates were extracted from HepG2 cells at 48 or 72 h post-transfection with HDAC5-shRNA. Then, Western blot was performed to determine the expression levels of apoptosis-related proteins (including p53, bcl-2, bax, cyto C, and caspases 3) and cell cycle-related proteins (such as p21, cyclin D1, and CDK2/4/6). β-actin was used as the internal control
HDAC5 knockdown promoted the stability, nuclear localization, and activity of p53. As a short-lived protein, p53 is degraded quickly under normal physiologic status. Therefore, enhancement of protein stability and nuclear localization is important for p53 to perform its transcriptional activity. We treated cells with CHX to block the protein synthesis in HepG2 cells, and Western blot showed that the down-regulation of HDAC5 significantly extended the half-life of p53 from 60 min to nearly 120 min (Fig. 6a). We further confirmed the localization of p53 by cellular fractionation and found that HDAC5-shRNA treatment for 48 or 72 h slightly promoted the accumulation of p53 in the cytoplasm, but significantly increased the localization of p53 in the nucleus (Fig. 6b). In addition, luciferase assay showed that the transactivation ability of p53 was significantly enhanced more than fourfolds after the down-regulation of HDAC5 (Fig. 6c). Taken together, these data demonstrate that knockdown of HDAC5 potentiates p53 function in HCC cells.

HDAC5 knockdown promoted p53 stability, nuclear localization, and activity. a HepG2 cells were transfected with HDAC5-shRNA and control shRNA for 72 h, and 20 μg/ml CHX was added to the medium. The cells were harvested at 0, 30, 60, 90, and 120 min. p53 expression was determined by Western blot and was quantified by densitometry. b HepG2 cells were transfected with HDAC5-shRNA and control shRNA for 48 or 72 h, and the p53 in cytoplasm and nucleus was detected using anti-p53, β-actin, and lamin B antibodies for Western blot. c HepG2 cells were transfected with the indicated plasmids, and luciferase reporter assays were performed. The relative luciferase activity was normalized to the value of Renilla activity. *P < 0.05. **P < 0.01
Discussion
It has been demonstrated that many members of the HDAC family play crucial roles in tumor initiation and development. For instance, class I HDACs can promote cancer cell proliferation and induce cell cycle arrest [18]. HDAC6, one member of class II HDACs, can alter cell motility and regulate the cytoskeleton [19]. Therefore, HDACs are regarded as promising targets for anti-cancer therapeutics.
HDAC5 belongs to the class II histone deacetylase family and is a critical regulator in cell proliferation in many cancer cell lines [20, 21]. A recent study found that HDAC5 was significantly overexpressed in high-risk medulloblastoma in comparison with low-risk medulloblastoma, and its expression was associated with poor survival, suggesting that HDAC5 may be an important marker for risk stratification [15]. Our present study found that HDAC5 was significantly increased in human HCC samples and cell lines. In addition, in vitro study showed that the down-regulation of HDAC5 by short hairpin RNA inhibited cell proliferation in HepG2, Hep3B, and Huh7 cells. Data from in vivo xenograft tumorigenesis model also showed the anti-proliferative effect of HDAC5, implicating the HDAC5 as a positive regulator in HCC cells.
Apoptosis is the process of programmed cell death characterized by typical cellular and molecular features such as cell shrinkage, externalization of phosphatidylserine, and condensation of chromatin [22, 23]. A recent study demonstrated that specific depletion of HDAC5 in cancer cells by RNA interference resulted in a significant change in heterochromatin structure, which triggered cancer cells to apoptosis, and arrested their growth both in vitro and in vivo [21]. In the present study, we found that the down-regulation of HDAC5 significantly increased cell apoptosis, indicating the possible mechanism by which knockdown of HDAC5 inhibited the proliferation of HCC cells. Cell cycle is a precise process controlled by cell cycle checkpoint which ensures the fidelity of cell division in all kinds of cells. However, cell cycle arrest could be triggered by various stimulating factors and resulted in the breakdown of cell division, cell death, and/or apoptosis [24–26]. Investigation of HDAC5 showed that it could transport from the nucleus to the cytoplasm and suppress the expression of cell cycle activator cyclin D, implicating its function in cell differentiation and proliferation [27]. Another study demonstrated that the chromatin structural defect caused by HDAC5 depletion resulted in a slow-growth phenotype, with delayed cell cycle progression and activation of multiple checkpoint pathways [21]. Not surprisingly, our study showed that the down-regulation of HDAC5 could induce G1-phase cell cycle arrest in the cell cycle progression.
Next, we explored the molecular mechanism underlying the anti-tumor effect by the determination of apoptosis- and cell cycle-related protein expression. Dysregulation of HDACs causes aberrant gene expression, thus promoting tumor development [28]. For this reason, the HDAC family is being studied as a molecular target for cancer therapeutics. At the molecular level, HDAC inhibitors have been linked to the induction of apoptosis and cell cycle regulation through the regulation of key cell growth genes like p53 and p21 [29, 30]. By means of such pathway, the inhibitors of HDAC family members selectively target tumorigenic cells both in vitro and in vivo [31, 32]. p53 plays a crucial role in multi-cellular organisms, regulating cell apoptosis and cell cycle [33]. We found that HDAC5 down-regulation resulted in a significant up-regulation of cyto C, caspase 3, p53 as well as its target gene bax. Moreover, the anti-apoptotic proteins bcl-2 was reduced in HDAC5-shRNA-transfected cells. The p21 protein acts as an inhibitor of cell cycle progression and negatively regulates the G1/S phase transition [34, 35]. Our results showed that the protein expression levels of cyclin D1 and CDK2/4/6 were significantly decreased, while p21 expression was significantly increased. Collectively, the changes of these proteins partially account for the induction of apoptosis and G1-phase cell cycle arrest by the down-regulation of HDAC5.
As an important transcriptional factor, p53 stability and nuclear localization are essential for its tumor suppressor function [36, 37]. We found that knockdown of HDAC5 significantly prolonged the half-life of p53 and induced its nuclear accumulation. Furthermore, we hypothesized that p53 stabilization and nuclear accumulation induced by HDAC5 depletion might increase its transcriptional activity. As expected, we found that the p53 transcriptional activity was significantly increased in HDAC5-shRNA-transfected cells.
In conclusion, our results for the first time demonstrated that the suppression of HDAC5 inhibited the proliferation of HCC cells through the induction of cell cycle arrest and apoptosis. These results not only provide further insight into the pathogenic mechanisms of HCC but also suggest HDAC5 as a potential target of future molecular therapies.
References
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27.
Yang Y, Jin C, Li H, et al. Improved radiosensitizing effect of the combination of etanidazole and paclitaxel for hepatocellular carcinoma in vivo. Exp Ther Med. 2012;3:299–303.
Zhang C, Ling Y, Zhang C, et al. The silencing of reck gene is associated with promoter hypermethylation and poor survival in hepatocellular carcinoma. Int J Biol Sci. 2012;8:451–8.
Minagawa M, Makuuchi M, Takayama T, et al. Selection criteria for repeat hepatectomy in patients with recurrent hepatocellular carcinoma. Ann Surg. 2003;238:703–10.
Johnson FM, Saigal B, Talpaz M, et al. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–32.
Yan K, Zhang C, Feng J, et al. Induction of G1 cell cycle arrest and apoptosis by berberine in bladder cancer cells. Eur J Pharmacol. 2011;661:1–7.
West AC, Mattarollo SR, Shortt J, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013;73:7265–76.
Sebastian C, Zwaans BM, Silberman DM, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–99.
Mottet D, Pirotte S, Lamour V, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–56.
Wilson AJ, Byun DS, Nasser S, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75.
Zhu C, Chen Q, Xie Z, et al. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: regulation on c-Myc. J Mol Med (Berl). 2011;89:279–89.
Wagner JM, Hackanson B, Lubbert M, et al. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–36.
Zhang Y, Matkovich SJ, Duan X, et al. Receptor-independent protein kinase c alpha (PKCalpha) signaling by calpain-generated free catalytic domains induces HDAC5 nuclear export and regulates cardiac transcription. J Biol Chem. 2011;286:26943–51.
Osada H, Tatematsu Y, Saito H, et al. Reduced expression of class ii histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004;112:26–32.
Milde T, Oehme I, Korshunov A, et al. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16:3240–52.
Lachenmayer A, Toffanin S, Cabellos L, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the hdac inhibitor panobinostat with sorafenib. J Hepatol. 2012;56:1343–50.
Liu H, Li P, Zhai Y, et al. Diagnostic value of glypican-3 in serum and liver for primary hepatocellular carcinoma. World J Gastroenterol. 2010;16:4410–5.
Glaser KB, Li J, Staver MJ, et al. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun. 2003;310:529–36.
Valenzuela-Fernandez A, Cabrero JR, Serrador JM, et al. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–7.
Lee CK, Wang S, Huang X, et al. HDAC inhibition synergistically enhances alkylator-induced dna damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010;296:233–40.
Peixoto P, Castronovo V, Matheus N, et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012;19:1239–52.
La Vignera S, Condorelli R, Vicari E, et al. Effects of varicocelectomy on sperm DNA fragmentation, mitochondrial function, chromatin condensation, and apoptosis. J Androl. 2012;33:389–96.
Ren W, Beebe SJ. An apoptosis targeted stimulus with nanosecond pulsed electric fields (nsPEFs) in E4 squamous cell carcinoma. Apoptosis. 2011;16:382–93.
Weinert T, Hopper AK. tRNA traffic meets a cell-cycle checkpoint. Cell. 2007;131:838–40.
Bai Y, Mao QQ, Qin J, et al. Resveratrol induces apoptosis and cell cycle arrest of human T24 bladder cancer cells in vitro and inhibits tumor growth in vivo. Cancer Sci. 2010;101:488–93.
Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(535–43):1–143.
McKinsey TA, Zhang CL, Lu J, et al. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–11.
Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25.
Richon VM, Sandhoff TW, Rifkind RA, et al. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–9.
Zhao Y, Tan J, Zhuang L, et al. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A. 2005;102:16090–5.
Kramer OH, Gottlicher M, Heinzel T. Histone deacetylase as a therapeutic target. Trends Endocrinol Metab. 2001;12:294–300.
Marks PA, Richon VM, Breslow R, et al. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–83.
Hu W, Ge Y, Ojcius DM, et al. P53 signalling controls cell cycle arrest and caspase-independent apoptosis in macrophages infected with pathogenic leptospira species. Cell Microbiol. 2013;15:1642–59.
Qiao D, Meyer K, Friedl A. Glypican-1 stimulates Skp2 autoinduction loop and G1/S transition in endothelial cells. J Biol Chem. 2012;287:5898–909.
Abe K, Naruse C, Kato T, et al. Loss of heterochromatin protein 1 gamma reduces the number of primordial germ cells via impaired cell cycle progression in mice. Biol Reprod. 2011;85:1013–24.
Jin L, Li C, Xu Y, et al. Epigallocatechin gallate promotes p53 accumulation and activity via the inhibition of MDM2-mediated p53 ubiquitination in human lung cancer cells. Oncol Rep. 2013;29:1983–90.
Yuan J, Luo K, Zhang L, et al. Usp10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–96.
Acknowledgments
This research was supported by the State Major Science and Technology Special Projects during the period of China State 12th 5-year plan (grant 2012ZX10002).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Jian Fan and Bin Lou contributed equally to this work.
Rights and permissions
About this article

Cite this article
Fan, J., Lou, B., Chen, W. et al. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumor Biol. 35, 11523–11532 (2014). https://doi.org/10.1007/s13277-014-2358-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2358-2