
Abstract
Patients undergoing androgen blockade therapy develop castration-resistant prostate cancer (CRPC), which is associated with Bcl-2 upregulation and results in disease progression and death. In recent years, promising therapeutic agents, such as the BH3-only mimetic ABT-263 and proteasome inhibitors, have been developed and widely evaluated against a broad spectrum of cancer types, including prostate cancer, alone or in combination with other chemotherapeutic agents. In this study, the antitumor efficacy of ABT-263 and MLN2238 were evaluated as single agents and in combination in four CRPC cell lines: PC3, C4-2B, C4-2, and DU145. The viability of the treated cells and markers of apoptosis were assayed. Protein-protein interactions were analyzed by co-immunoprecipitation in drug-treated cells. Lentivirus-mediated short hairpin RNA was used to knockdown Bax, Mcl-1, and NOXA expressions. We found that ABT-263 and MLN2238 alone exhibited a mild cytotoxicity, and in combination, they elicited a synergistic cytotoxic effect in CRPC cells. The cell apoptosis induced by the combination drug treatment was evidenced by enhanced caspase-3 and Poly (ADP-ribose) polymerase (PARP) cleavage, and annexin-V-positive staining was significantly depleted by Bax knockdown. MLN2238 treatment upregulated NOXA and Mcl-1 expression, leading NOXA/Mcl-1 complexes to disassociate Bak from its complexes with Mcl-1 and enhancing ABT263-triggered Bax activation. NOXA knockdown by short hairpin RNA significantly attenuated the cytotoxicity of ABT-263 and MLN2238 co-administration. In conclusion, MLN2238 and ABT-263 synergistically triggered apoptosis in CRPC cells by upregulating NOXA and activating Bax, indicating a promising therapeutic strategy for the treatment of CRPC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is one of the most commonly diagnosed solid tumors in men in the developed world and accounts for the second leading cause of cancer-related deaths in North American men. Androgens play a key role in promoting the development and progression of prostate cancer. Androgen deprivation therapy (ADT) is generally used in prostate cancer patients [1, 2]. However, most patients with ADT usually develop castration-resistant prostate cancer (CRPC) within 2 years, which results in disease progression and mortality. Although researchers have developed therapeutic strategies to sensitize ADT that show promise for clinical applications in future trials [3–5], there is still a need to develop novel therapeutics to improve the survival and quality of life in patients with androgen-independent and advanced prostate cancer.
The mechanisms underlying the resistance to ADT are still not well understood. More and more pieces of evidence show that the modulation of Bcl-2 family members might trigger apoptosis and enhance chemotherapies in CRPC cells [6, 7]. Bcl-2 family proteins include more than 20 members. They are mainly divided into two groups: pro-survival members (e.g., Bcl-2, Bcl-XL, Bcl-w, Mcl-1, and A1) and pro-apoptotic members (e.g., Bax, Bak, NOXA, and Bim). Their interaction and dissociation govern the mitochondrial outer membrane permeabilization (MOMP) that results in the release of apoptotic factors (e.g., cytochrome c) and the subsequent activation of caspases [8]. A constitutively high level of pro-survival proteins, such as Bcl-2, has been correlated with high rates of cancer recurrence and chemotherapy resistance in patients with primary prostate cancer [9]. Therefore, modulating Bcl-2 family members may be an effective therapeutic strategy to trigger programmed cell death in many cancer types, including prostate cancer.
ABT-263/ABT-737, discovered by Abbott Laboratories, are potent small molecules with a high binding affinity (nM range) to Bcl-2, Bcl-XL, and Bcl-w but not Mcl-1 or A1 [10]. ABT-263 acts like a BH3-only protein to neutralize the pro-survival members and thereby diminish their ability to inhibit cell apoptosis. Currently, ABT-263 is widely studied in preclinical research and clinical trials against various cancer types, including prostate cancer. Although studies have shown that long-term treatment with ABT-263/ABT-737 usually results in the upregulation of Mcl-1, which is a main reason for acquired drug resistance [11, 12], ABT-263/ABT-737 is still a good chemopotentiator to enhance other chemotherapeutic agents against multiple types of cancers [13–15]. The ubiquitin proteasome pathway plays a critical role in regulating tumor cell growth and survival. Blocking the proteasome’s function by proteasome inhibitors has proven to be a powerful strategy for cancer therapy [16, 17]. MLN2238, the hydrolized form of MLN9708, is an orally active proteasome inhibitor. It is a next-generation proteasome inhibitor. Compared with bortezomib, MLN9708/MLN2238 has a shorter proteasome dissociation half-life, which may improve its tissue distribution [18]. Studies have shown that MLN9708/MLN2238 displays a strong antitumor effect on a broad spectrum of tumor cell lines in vitro and is efficacious against human prostate xenografts in vivo [18, 19].
Given the rationale that different classes of drugs may complement each other through different effects to trigger the apoptotic cascade, in this study, we investigated the antitumor efficacy of the BH3-only mimetic ABT-263 and MLN2238 alone and in combination on CRPC cell lines. Our data indicate that MLN2238 sensitizes ABT-263-induced cytotoxicity by upregulating NOXA expression and triggering Bax activation.
Materials and methods
Cell lines and reagents
The human castration-resistant prostate cancer cell lines PC3 and DU145 (ATCC, Manassas, VA, USA), C4-2 (Urocor, Oklahoma City, OK), and C4-2B (ViroMed Laboratories, Minnetonka, MN) were cultured in RPMI 1640 containing 10 % fetal bovine serum with 1 % penicillin/streptomycin in a humidified atmosphere of 5 % CO2 at 37 °C. ABT-263 and MLN2238 were purchased from ChemieTek (Indianapolis, IN). The compounds were dissolved in DMSO to produce 10-mmol/L stock solutions, and the aliquots were stored at −80 °C for usage.
Cell viability analysis
The cells were seeded into 24-well plates for the cell viability analysis. After overnight culture, the cells were exposed to the compounds as indicated. After incubation, the cells were harvested and stained with trypan blue. The cells’ viabilities were determined with the TC10™ Automatic Cell Counter (Bio-Rad, Hercules, CA). Each experiment was performed in triplicate, at least three times.
Measurement of cellular apoptosis
The cells in six-well plates were treated with ABT-263 and MLN2238, alone or in combination, as indicated for 1 day. Floating and adherent cells were harvested and stained with propidium iodide and FITC-annexin V according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). The stained cells were analyzed by flow cytometry (BD FACSCanto™ II Analyzer, BD Biosciense, San Jose, CA) to quantitate the annexin-V-positive apoptotic cells.
Short hairpin RNA
The pGreenPuro short hairpin RNA (shRNA) vector was purchased from System Biosciences (Mountain View, CA). The shRNA lentivirus plasmids were made by cloning specific target oligonucleotides into the lentiviral vector according to the manufacturer’s instruction. The target sequences of shRNA template oligonucleotide used were as follows: sh-Mcl-1, 5′-GGACTTTTATACCTGTTAT-3′, and sh-NOXA, 5′-GTAATTATTGACACATTTCTT-3′. The details of the control shRNA and sh-Bax were described previously [20]. The viruses were produced by co-transfecting 293TN cells with a lentiviral vector, packaging, and envelope plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The virus-containing supernatants were harvested 48- and 72-h post-transfection, then either used for infection or stored in aliquots at −80 °C.
Immunoprecipitation
Cells in six-well plate treated with drugs were harvested by scraping and then washed in cold PBS once. After collection, cells were lysed with 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) buffer (20 mmol/L Tris–HCl, pH 7.5; 150 mmol/L NaCl; 1 mmol/L EDTA; 2 % CHAPS) supplemented with protease inhibitor cocktail for 1 h on ice. Protein lysates were incubated with primary antibody of Bax (6A7) (Sigma, St. Louis, MO), Mcl-1 (BD-Biosciences, San Jose, CA), and Bak (Cell Signaling Technologies, MA, US) overnight at 4 °C followed by 2 h of incubation of protein A agarose beads. After three washes with lysis buffer, proteins were recovered by boiling the beads in sodium dodecyl sulfate (SDS) sample buffer and analyzed by Western blotting analysis. Each experiment was performed at least three times.
Immunoblotting
Floating and adherent cells were harvested and prepared with 1 % NP-40 lysis buffer (20 mmol/L Tris–HCl, pH7.5; 150 mmol/L NaCl; 1 mmol/L EDTA; 1 % NP-40) containing protease inhibitors (Roche). Then, 50-μg cell lysates were separated on SDS-PAGE electrophoresis and transferred to PVDF membrane (Bio-Rad, Hercules, CA). Blots were probed with polyclonal or monoclonal antibodies against Mcl-1(BD-Biosciences, San Jose, CA), Bim (Cell Signaling Technologies, MA, US), NOXA, Bax, Bcl-2, Bcl-XL, Bcl-w, Bak, and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
Statistical analysis
Data from studies were expressed as the mean ± standard deviation. The statistical significances of differences between groups were determined using Student’s t test. Difference was considered statistically significant if p < 0.05.
Results
MLN2238 synergistically enhanced ABT-263-induced cytotoxicity in castration-resistant prostate cancer cells
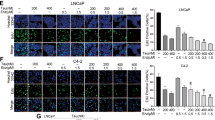
Previous studies have shown that BH3 mimics of ABT-737 could synergistically enhance the cancer cell death induced by the first-generation proteasome inhibitor, bortezomib, in human melanoma cancer cells and lymphoid malignancies [21, 22]. MLN2238 is an orally active proteasome inhibitor, which has improved pharmacokinetic and pharmacodynamic profiles compared to bortezomib [18]. Here, we start to analyze the interaction between ABT-263 and MLN2238 in PC3 cells in vitro. As shown in Fig. 1a, PC3 cells were given a fixed concentration of MLN2238 (75 nM) in the presence or absence of increasing dosages of ABT-263 (250 to 1,000 nM) for 1 day. ABT-263 and MLN2238 treatment alone only showed minimal cytotoxicity (approximately 8 % cell death with 75 nM MLN2238 or 1,000 nM ABT-263), while the co-administration of ABT-263 and MLN2238 significantly increased cell death as the concentration of ABT-263 increased (approximately by 68.3 % at 1,000 nM, p < 0.01). Similarly, when PC3 cells were exposed to increasing doses of MLN2238 (25 to 100 nM) in the presence or absence of ABT-263 (1,000 nM), MLN2238 significantly sensitized PC3 cells to ABT-263-induced cytotoxicity, also in a dose-dependent manner (Fig. 1b). In addition, PC3 cells were co-treated with fixed concentration of ABT-263 (500 nM) and MLN2238 (50 nM) for up to 3 days. As shown in Fig. 1c, the combination treatment killed cells in a time-dependent manner. The synergistic interaction efficacy of ABT-263 and MLN2238 was analyzed using the method of Chou and Talalay [23]. PC3 cells were exposed to different concentrations of ABT-263 and MLN2238 at a fixed ratio (10:1), and the combination index (CI) was calculated with the CalcuSyn program. The values of CI lower than 1.0 indicated a synergistic interaction (Fig. 1d). Moreover, the co-administration of ABT-263 and MLN2238 significantly induced more cell death than either drug alone in the CRPC cell lines C4-2B, C4-2, and DU145 (Fig. 1e), indicating that MLN2238 synergistically sensitized CRPC cells to ABT-263-induced antitumor efficacy.

MLN2238 synergistically enhanced ABT-263-induced cytotoxicity in androgen-independent prostate cancer cells. PC3 cells were treated with a MLN2238 (MLN) in the presence of the indicated dose of ABT-263 (ABT) or b with fixed concentration of ABT-263 and the indicated concentration of MLN2238 for 1 day; the cell death rates were analyzed as described in the “Materials and methods” section. c PC3 cells were exposed to ABT-263 (500 nM) and MLN2238 (50 nM), alone or in combination, for the indicated time. Cell viabilities were determined. The data are shown as the mean ± standard deviation. d Combination indices (CI) of synergistic interactions between ABT-263 and MLN2238 with a fixed ratio of 10:1 were calculated with the CalcuSyn program. CI values lower than 1.0 indicate synergism. e The PC3, C4-2B, C4-2, and DU145 androgen-independent prostate cancer cell lines were treated for 1 day with ABT-263 (1,000 nM), MLN2238 (75 nM), or both, and cell death rates were determined. For this and the following figures, statistical significance is indicated by asterisk if p < 0.05 or if **p < 0.01
Co-treatment with MLN2238 and ABT-263 resulted in mitochondrial pathway-dependent apoptosis
To analyze whether the antitumor efficacy caused by co-treatment with MLN2238 and ABT-263 was the result of cell apoptosis, PC3 and C4-2B cells were treated with MLN2238 and ABT-263, either alone or in combination, and were harvested for immunoblotting analysis. As shown in Fig. 2a, the co-treatment with ABT-263 and MLN2238 resulted in the cleavage of caspase-3 and Poly (ADP-ribose) polymerase (PARP), which are markers of programmed cell death. The cell deaths induced by the combination treatment in PC3 and C4-2B cells were further shown to be related to apoptosis with annexin V staining. The drug combination markedly triggered more apoptosis than either drug alone did (Fig. 2b). To test whether combination treatment-triggered apoptosis is mitochondrial-pathway-dependent, Bax was knocked down by lentivirus-mediated short hairpin RNA in PC3 cells. As expected, the knockdown of Bax significantly protected PC3 cells from ABT-263 and MLN2238 co-treatment-induced apoptosis (Fig. 2c).

Co-treatment with ABT-263 and MLN2238 triggers the intrinsic apoptotic pathway. a PC3 and C4-2B cells were treated with ABT-263 (1,000 nM), MLN2238 (75 nM), or both for 1 day, and then cleaved caspase-3 and PARP were detected by Western blotting. β-Actin was determined as an endogenous control. b PC3 and C4-2B cells were treated as described in a. Cellular apoptosis was determined by flow cytometry using propidium iodide and FITC-annexin-V staining. c Bax expression in PC3 cells was knocked down by lentivirus-mediated short hairpin RNA (shRNA) (sh-Bax). Cells were then treated with ABT-263, MLN2238, or both, as in a, followed by Western blotting analysis of the cleavage of caspase-3 and PARP (left) and apoptotic cell death analysis (right)
MLN2238 treatment resulted in NOXA and Mcl-1 upregulation
To determine whether MLN2238 modulates Bcl-2 family members and then enhances the cytotoxicity induced by the BH3 mimetic, ABT-263, PC3, and C4-2B cells were exposed to increasing doses of MLN2238 for 1 day. As shown in Fig. 3a, the pro-apoptotic protein NOXA and the pro-survival protein Mcl-1 were upregulated significantly, especially at dosages of 50 and 100 nM. There were no obvious changes detected with other Bcl-2 family members, including Bcl-2, Bcl-XL, Bcl-w, and Bax. When cells were cultured in the presence of 50 nM MLN2238 for up to 24 h, NOXA and Mcl-1 were induced robustly in a time-dependent manner, especially after 8 h of treatment (Fig. 3b). Meanwhile, we found that the upregulation of NOXA by MLN2238 is stronger than that of Mcl-1, e.g., a 44-fold upregulation of NOXA versus an 8.8-fold increase of Mcl-1 in PC3 cells in the presence of 50 nM MLN2238 for 24 h (Fig. 3b). Although it has been reported that proteasome inhibition can lead to an upregulation of Bim [24], here, Bim expression was little affected by MLN2238 (Fig. 3c). We then investigated the modulation of NOXA and Mcl-1 in the combination treatment condition. As shown in Fig. 4a, NOXA expression was robustly upregulated after treatment with MLN2238 alone or in combination with ABT-263. Mcl-1 was upregulated by treatment with ABT-263 or MLN2238 alone. Interestingly, there was no further upregulation with the co-treatment of ABT-263 and MLN2238. Instead, the Mcl-1 protein level was a little lower after the combination treatment than after the MLN2238-alone treatment (Fig. 4a).

MLN2238 treatment resulted in the upregulation of NOXA and Mcl-1. a PC3 and C4-2B cells were treated with increasing concentrations of MLN2238 for 24 h; then, Bcl-2 family members were analyzed by Western blotting. b PC3 and C4-2B cells were treated with MLN2238 (50 nM) for up to 24 h. NOXA, Mcl-1, and Bim expressions were examined by immunoblotting. The intensity of the NOXA and Mcl-1 signals was quantified by densitometry using ImageJ 1.47 t software and normalized to 0 nM in dose-response and 0 h in time-response detection, which is shown below the immunoblot results

NOXA upregulation by MLN2238 neutralizes Mcl-1 and results in Bax activation. a PC3 and C4-2B cells were exposed to ABT-263 (1,000 nM), MLN2238 (75 nM), or both for 1 day before the cells were harvested for the immunoprecipitation of Mcl-1 to examine the interaction of Mcl-1 and NOXA by co-immunoprecipitation. b The interactions of Bak and Mcl-1 or Bak and Bcl-XL were determined by co-immunoprecipitation of Bak. c C4-2B cells were treated for 1 day with ABT-263 (1,000 nM), MLN2238 (75 nM), or both and lysed in 2 % CHAPS buffer, followed by immunoprecipitation and Western blot analysis of active Bax
NOXA upregulation by MLN2238 sequestered Mcl-1 and resulted in Bax activation
Bcl-2 family members are essential players in the control of the intrinsic apoptosis pathway. Their selective binding and disassociation regulate Bax and Bak activation and mitochondrial outer membrane permeabilization. Mcl-1 is a pro-survival factor, which is recognized by the BH3-only protein NOXA and the multi-BH protein Bak [8]. Given the upregulation of NOXA and Mcl-1 by MLN2238 in PC3 and C4-2B cells, we investigated the effects of drug treatment on the interactions between Mcl-1 and its interaction partners. To address this issue, PC3 and C4-2B cells were treated with ABT-263 and MLN2238 alone or in combination before the cells were harvested for immunoprecipitation by Mcl-1. NOXA and Bak were probed. As shown in Fig. 4a, ABT-263 treatment had little effect on the Mcl-1 and NOXA interaction, whilst MLN2238 was shown to robustly enhance this interaction. The upregulation of NOXA could activate Bak by competitively releasing it from Mcl-1 or Bcl-XL [25]. As expected, MLN2238 disrupted Bak from Mcl-1 and Bcl-XL (Fig. 4b). We then analyzed the activation of Bax by immunoprecipitation with the 6A7 antibody, which can recognize the N terminus of the activated form of Bax [26]. As shown in Fig. 4c, co-treatment with ABT-263 and MLN2238 caused Bax activation. Taken together, our data show that the upregulation of NOXA by MLN2238 sequestered Mcl-1 and disrupted Bak from its partners, which may be the key to its synergism with ABT-263 in androgen-independent prostate cancer cells.
Depletion of NOXA protects PC3 cells from drug combination-induced cytotoxicity
Mcl-1 upregulation was reported as a main reason for acquired drug resistance [11, 12]; however, we found that the knockdown of Mcl-1 significantly enhanced ABT-263-induced cell death, shown as increased caspase-3 and PARP cleavage (Fig. 5a). Moreover, given that NOXA was induced by MLN2238, we then investigated whether NOXA upregulation contributed to the drug combination-induced cell apoptosis. As shown in Fig. 5b, NOXA induction was significantly inhibited by lentivirus-mediated shRNA. NOXA knockdown markedly protected PC3 cells from ABT-263 and MLN2238-triggered cell apoptosis, as evidenced by the blocked activation of caspase-3 (upper panel) and decreased apoptotic cell death (from 62 % with control shRNA to 24 % with sh-NOXA). Together, those data indicate that NOXA is a key player in combination-induced antitumor efficacy.

Depletion of NOXA protected PC3 cells from ABT-263 and MLN2238-induced apoptosis. a Mcl-1 expression in PC3 cells was downregulated by lentivirus-mediated shRNA. The Mcl-1 knockdown cells (sh-Mcl-1) and the control shRNA-infected cells (sh-ctrl) were treated for 1 day with ABT-263 (1,000 nM). The cells were harvested for Western blot analysis of Mcl-1 and the cleavage of caspase-3 and PARP (left) or cell death analysis (right). b NOXA expression in PC3 cells was knocked down by shRNA. The NOXA knockdown cells (sh-NOXA) and control shRNA-infected cells (sh-ctrl) were treated for 1 day with ABT-263 (1,000 nM) and MLN2238 (75 nM), alone or in combination, followed by immunoblotting for NOXA and cleaved caspase-3 (upper) and cell death analysis (lower)
Discussion
Castration-resistant prostate cancer is a devastating disease, as it resists all conventional treatments, resulting in cancer progression and death. A number of genetic abnormalities in androgen receptors and Bcl-2 family proteins are likely involved in this progression [27, 28]. In this study, we investigated whether the strategy of combining the BH3-only mimetic ABT-263 and the second generation proteasome inhibitor MLN2238 could synergistically affect cell regression in CRPC cells. We found that the co-administration of ABT-263 and MLN2238 synergistically caused cytotoxicity in CRPC cells due to apoptosis, shown by the marked cleavage of apoptotic markers and the increase of annexin-V-positive staining. And, the knockdown of Bax protected PC3 cells from the combination treatment further supported this point (Figs. 1 and 2). Furthermore, we showed that MLN2238 treatment resulted in a robust upregulation of NOXA, which sequestered Mcl-1from its antiapoptotic functions and enhanced ABT-263-triggered apoptosis in CRPC cells (Figs. 3, 4, and 5), suggesting the combination treatment as a promising therapeutic method to treat CRPC.
The upregulation of the pro-survival protein Bcl-2 is required for the progression of CRPC growth [28]. Targeting Bcl-2 is an attractive way to cause cancer cell regression in CRPC [29–31]. The BH3 mimetic ABT-263/ABT-737 was widely used in the treatment of lymphomas and solid tumors in clinical and preclinical studies to sequester Bcl-2/BCL-XL/Bcl-w [11]. ABT-263/ABT-737 as a single treatment agent has limited anticancer effects due to its inactive binding to Mcl-1. The inhibition of Mcl-1 was proven to be an effective strategy to enhance ABT-263/ABT-737 functions. As we observed, Mcl-1 knockdown significantly enhanced ABT-263-induced cell death in PC3 cells (Fig. 5a).
Similar to BH3 mimetics, proteasome inhibitors are another type of anticancer agent; drugs of this class modulate pro-apoptotic factors, such as p53 and NOXA, and induce apoptosis in numerous malignant cell types when used as a single agent or in combination with other chemotherapeutic agents. As reported, Celastrol, an active compound from Chinese medicine, also called “Thunder of God Vine,” is a natural proteasome inhibitor with potential for prostate cancer treatment [32]. Furthermore, the proteasome inhibitor PS-341 even killed prostate cancer cells grown in the form of multicellular spheroids [33].
The second-generation proteasome inhibitor MLN2238/MLN9708 is currently under evaluation in a phase I clinical trial [19]. It is known that the combination of first-generation proteasome inhibitor MG-341, or bortezomib, and ABT-737 triggers cell apoptosis in human melanoma and lymphoid malignancies [21, 22]. In our study, we found that MLN2238 significantly induced NOXA expression in a time- and dose-dependent manner in PC3 and C4-2B cells. The induction of Mcl-1 by ABT-263 was regarded as the main reason for acquired drug resistance and a major obstacle to chemotherapeutic induction of apoptosis. Interestingly, although an upregulation of Mcl-1 expression was observed in the cases of ABT-263 and MLN2238 treatment alone, its expression was decreased in the combination treatment. As a BH3-only protein, NOXA interacts only with Mcl-1 and A1, which are not able to be sequestered by ABT-263. As we observed, although Mcl-1 was upregulated after MLN2238 treatment, the NOXA/Mcl-1 protein ratio increased in a time- and dose-dependent manner (Fig. 3). The enhancement of NOXA and Mcl-1 interactions by MLN2238 is highly correlated with this finding. Additionally, the NOXA/Mcl-1 ratio was shown to control cell susceptibility to chemodrug-induced apoptosis [34].
Together, our data indicate that NOXA plays an important role in the synergism of MLN2238 and ABT-263 in releasing Bim and Bak by neutralizing Mcl-1 expression. To confirm the key role of NOXA in the synergistic activity of ABT-263 and MLN2238, NOXA downregulation by shRNA was shown to markedly decrease combination-treatment-triggered cell apoptosis and thus protect PC3 cells (Fig. 5).
In conclusion, our studies show that MLN2238 synergistically potentiated ABT-263-induced cytotoxicity in CRPC cells in vitro. MLN2238 induced NOXA expression. NOXA upregulation antagonized Mcl-1 induction by disrupting Mcl-1 from its interaction with Bak and triggered Bax activation. These findings validate the rational strategy to treat castration-resistant prostate cancer by combining the BH3 mimetic ABT-263 and the proteasome inhibitor MLN2238.
Abbreviations
- ADT:
-
Androgen deprivation therapy
- CI:
-
Combination index
- CRPC:
-
Castration-resistant prostate cancer
- MOMP:
-
Mitochondrial outer membrane permeabilization
References
Catalona WJ. Management of cancer of the prostate. N Engl J Med. 1994;331:996–1004.
Tammela T. Endocrine treatment of prostate cancer. J Steroid Biochem Mol Biol. 2004;92:287–95.
Chen L, Mooso BA, Jathal MK, Madhav A, Johnson SD, van Spyk E, et al. Dual EGFR/HER2 inhibition sensitizes prostate cancer cells to androgen withdrawal by suppressing ErbB3. Clin Cancer Res. 2011;17:6218–28.
Tovar C, Higgins B, Kolinsky K, Xia M, Packman K, Heimbrook DC, et al. MDM2 antagonists boost antitumor effect of androgen withdrawal: implications for therapy of prostate cancer. Mol Cancer. 2011;10:49.
Oh WK, Kantoff PW. Management of hormone refractory prostate cancer: current standards and future prospects. J Urol. 1998;160:1220–9.
Yamanaka K, Rocchi P, Miyake H, Fazli L, Vessella B, Zangemeister-Wittke U, et al. A novel antisense oligonucleotide inhibiting several antiapoptotic Bcl-2 family members induces apoptosis and enhances chemosensitivity in androgen-independent human prostate cancer PC3 cells. Mol Cancer Ther. 2005;4:1689–98.
Pandit B, Gartel AL. New potential anti-cancer agents synergize with bortezomib and ABT-737 against prostate cancer. Prostate. 2010;70:825–33.
Billard C. Design of novel BH3 mimetics for the treatment of chronic lymphocytic leukemia. Leukemia. 2012;26:2032–8.
Yoshino T, Shiina H, Urakami S, Kikuno N, Yoneda T, Shigeno K, et al. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin Cancer Res. 2006;12:6116–24.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8.
Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–13.
Mazumder S, Choudhary GS, Al-Harbi S, Almasan A. Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012;72:3069–79.
Mason KD, Khaw SL, Rayeroux KC, Chew E, Lee EF, Fairlie WD, et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–41.
Tang H, Shao H, Yu C, Hou J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochem Pharmacol. 2011;82:1066–72.
Li R, Zang Y, Li C, Patel NS, Grandis JR, Johnson DE. ABT-737 synergizes with chemotherapy to kill head and neck squamous cell carcinoma cells via a Noxa-mediated pathway. Mol Pharmacol. 2009;75:1231–9.
McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11:164–79.
Cusack JC. Rationale for the treatment of solid tumors with the proteasome inhibitor bortezomib. Cancer Treat Rev. 2003;29 Suppl 1:21–31.
Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–80.
Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–9.
Shao H, Gao C, Tang H, Zhang H, Roberts LR, Hylander BL, et al. Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer in vitro and in vivo. J Hepatol. 2012;56:176–83.
Miller LA, Goldstein NB, Johannes WU, Walton CH, Fujita M, Norris DA, et al. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J Invest Dermatol. 2009;129:964–71.
Paoluzzi L, Gonen M, Bhagat G, Furman RR, Gardner JR, Scotto L, et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 2008;112:2906–16.
Chou TC, Talalay P. Quantitative analysis of doseeffect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.
Westerberg CM, Hägglund H, Nilsson G. Proteasome inhibition upregulates Bim and induces caspase-3-dependent apoptosis in human mast cells expressing the Kit D816V mutation. Cell Death Dis. 2012;3:e417.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305.
Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999;18:2330–41.
Catz SD, Johnson JL. BCL-2 in prostate cancer: a minireview. Apoptosis. 2003;8:29–37.
Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM, Xiang J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007;17:531–6.
Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995;55:4438–45.
Gleave ME, Miayake H, Goldie J, Nelson C, Tolcher A. Targeting bcl-2 gene to delay androgen-independent progression and enhance chemosensitivity in prostate cancer using antisense bcl-2 oligodeoxynucleotides. Urology. 1999;54(6A Suppl):36–46.
Yamanaka K, Rocchi P, Miyake H, Fazli L, Vessella B, Zangemeister-Wittke U, et al. A novel antisense oligonucleotide inhibiting several antiapoptotic Bcl-2 family members induces apoptosis and enhances chemosensitivity in androgen-independent human prostate cancer PC3 cells. Mol Cancer Ther. 2005;4:1689–98.
Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine”, is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–65.
Frankel A, Man S, Elliott P, Adams J, Kerbel RS. Lack of multicellular drug resistance observed in human ovarian and prostate carcinoma treated with the proteasome inhibitor PS-341. Clin Cancer Res. 2000;6:3719–28.
Mei Y, Xie C, Xie W, Tian X, Li M, Wu M. Noxa/Mcl-1 balance regulates susceptibility of cells to camptothecin-induced apoptosis. Neoplasia. 2007;9:871–81.
Acknowledgments
This work was supported by the National Natural Science Foundation Grant of China (No. 81072091/H1619) and Key Project of Guangzhou Municipal Health Bureau Grant, China (No. 20121A021006) to Ping Tang.
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Xinghua Wei, Ping Zhou, and Xuanting Lin contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wei, X., Zhou, P., Lin, X. et al. MLN2238 synergizes BH3 mimetic ABT-263 in castration-resistant prostate cancer cells by induction of NOXA. Tumor Biol. 35, 10213–10221 (2014). https://doi.org/10.1007/s13277-014-2333-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2333-y