
Abstract
Hepatocellular carcinoma is the fifth most common tumor and the third cause of death for cancer in the world. Among the main causative agents of this tumor is the chronic infection by hepatitis viruses B and C, which establish a context of chronic inflammation degenerating in fibrosis, cirrhosis, and, finally, cancer. Recent findings, however, indicate that hepatitis viruses are not only responsible for cancer onset but also for its progression towards metastasis. Indeed, they are able to promote epithelial-mesenchymal transition, a process of cellular reprogramming underlying tumor spread. In this manuscript, we review the currently known molecular mechanisms by which hepatitis viruses induce epithelial-mesenchymal transition and, thus, hepatocellular carcinoma progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Epidemiology and pathogenesis of hepatocellular carcinoma
Hepatocellular carcinoma (HCC), being the most common subtype of liver cancer (70 to 85 %), is the fifth most frequent neoplasm, affecting more than half a million people each year worldwide [1]. The highest frequencies of HCC are found in developing countries (East and Southeast Asia and Middle and Western Africa), although the incidence is rising in developed western countries too [2–4]. These geographical differences are explained by both different prevalence of risk factors and ethnical factors [5].
In most cases, hepatocarcinogenesis arises in the setting of chronic liver damage and inflammation, which leads to fibrosis and, ultimately, to cirrhosis [6]. Among the main causes underlying chronic liver damage are toxicity from heavy alcohol consumption and from dietary aflatoxins, metabolic disorders like diabetes, non-alcoholic fatty liver disease, hereditary hemochromatosis, porphyria cutanea tarda, hereditary tyrosinemia, α1 anti-trypsin deficiency, and immunological disorders such as primary biliary cirrhosis and autoimmune hepatitis [3, 5]. The leading risk factor for HCC, however, is represented by chronic viral hepatitis. About 80 % of cases of HCC arise following chronic infection by hepatitis B virus (HBV) or hepatitis C virus (HCV) [7]. Some studies also report hepatitis D virus (HDV) infection as a potential risk factor for HCC [8–11].
Hepatitis viruses can induce HCC through different mechanisms, such as activation/inactivation of cellular genes due to viral genome integration, modulation of cell signaling and metabolic pathways, alteration of cell growth and apoptosis, transcriptional and translational regulation, epigenetic regulation, induction of oxidative stress, and modulation of the host immune response [5, 6, 12–15]. The two latter aspects play a major role in viral-induced hepatocarcinogenesis since viral infection elicits an immune response which often fails to clear the virus but, in turn, creates a continuous cycle of hepatocytes’ death and compensatory regeneration, inflammation, and tissue repair, which promote liver fibrosis, cirrhosis, and cancer [5, 6, 12, 13]. This chronic activation of the immune system driven by hepatitis viruses worsens the condition of oxidative stress by dysregulating cell signaling and metabolism [6]. Several reports indicate the existence of a link between hepatitis virus infection and oxidative damage: In humans, chronic infection of the liver is associated with increased content of 8-hydroxydeoxiguanosine, a DNA modification caused by reactive oxygen species (ROS) [16, 17], while in transgenic mice, expressing either HBV- or HCV-increased oxidative stress correlates with HCC development [16, 18–20]. Oxidative stress, in turn, activates the MAPK cascade and the hepatic stellate cells which promote cell proliferation and fibrogenesis [16] .
Surgical resection, liver transplantation, and local ablation are the main curative treatment options for hepatocellular carcinoma, even though therapies should be individualized depending on tumor stage, and only few patients with early stage disease are eligible for these therapeutic approaches [21, 22]. For advanced stage HCC patients, instead, the therapies are mainly palliative or symptomatic, apart from the treatment with sorafenib. The latter is a multikinase inhibitor of the vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and Raf, targeting cell proliferation, apoptosis, and angiogenesis [23]. The rationale for its approvement in the treatment of unresectable HCC relies on frequent activation of these tyrosine-kinases in HCC [24]. The response to sorafenib, however, is low, with less than 3 months of survival prolongation [24] and patients may develop resistance [25]; thus, the finding of further therapeutic approaches is urgently needed.
Due to the high vascularity of liver, HCC is prone to metastasize, mainly at the intrahepatic level and at the extrahepatic one, with lungs, bones, lymph nodes, and adrenal glands being most frequently involved [26]. Metastases are among the main causes of ineffectiveness of the current therapies, making HCC the third cause of cancer-related death in the world [27].
Epithelial-mesenchymal transition: a mechanism of cancer progression
A crucial mechanism in cancer progression and metastasis, which is known to be also directly induced by hepatitis viruses, is epithelial-mesenchymal transition (EMT).
EMT is a process in which epithelial cells undergo a transcriptional reprogramming and dramatic morphological changes and acquire a mesenchymal phenotype characterized by more migratory properties. Epithelial cells compose organized layers in which cells are adherent to each other: this is due to the arrangement of different kinds of cell-cell junctions (i.e., adherens junctions, desmosomes, and tight junctions) which limit their motility and prevent their detachment. Moreover, some protein complexes (such as Par and Crumbs) associate with the junctions and allow epithelial cells to acquire an apicobasal polarity [28]. On the contrary, mesenchymal cells are elongated in shape, not polarized, and lack intercellular interactions. This enables them to cross extracellular matrix and stromal tissues and to disseminate through the circulatory system [29–31].
At the molecular level, the changes occurring during EMT are explained by the loss of epithelial markers and the gain of mesenchymal ones. A major event in EMT is the downregulation of E-cadherin promoted by a number of transcription factors, among which are those belonging to the families of SNAIL, TWIST, and ZEB [32, 33]. Another mechanism by which E-cadherin is downregulated, as observed in different kinds of tumors, is the hypermethylation of its promoter induced by DNA methyltransferases (DNMTs) [34]. Other hallmarks of EMT are the increased expression of protein factors involved in re-organization of cytoskeleton and motility, such as FSP1, α-SMA and vimentin [33], the increased production of metalloproteinases for matrix degradation [35], and the cytoplasm-to-nucleus translocation of β-catenin, a protein that not only connects cadherins to cytoskeleton but also regulates gene expression associated with EMT [33]. Once in the nucleus, β-catenin binds to the high-mobility group domain factors Tcf/LEF and activates the expression of some mesenchymal markers such as fibronectin and matrix metalloproteases [36, 37], some repressors of epithelial differentiation, such as CDX1 and ENC1, beside a number of genes regulating cell proliferation, survival, and invasiveness [37].
A mediator of EMT is represented by ROS, which are known to play a role in the hepatocarcinogenesis, as stated above. Indeed, ROS can activate the MAPK cascade, thus supporting proliferation; they can regulate transcription factors involved in tumor progression (including Snail) and genes promoting invasion (such as metalloproteases); and they can induce cytoskeletal remodeling, an event which takes place in EMT [38].
Based on the biological context in which it takes place, EMT can be classified into three subtypes. Type 1 EMT physiologically occurs during embryo implantation, embryo development, and organogenesis. Type 2 EMT occurs both in the physiological response to injury during wound healing and tissue repair and in the pathological setting of organ fibrosis. Type 3 EMT is a process associated with metastatic cancer: it allows neoplastic cells to become motile and invasive and leave the primary epithelial tumor site to reach, through the bloodstream, different body districts where they form a secondary tumor [28, 30, 39]. Since after the colonization the secondary tumor cells resemble the tumor from which they originate and no longer show mesenchymal features, the reverse process is thought to occur, namely mesenchymal-epithelial transition (MET) in which mesenchymal cells revert to an epithelial phenotype [39, 40].
A growing body of literature reports a pivotal role for EMT in the invasiveness of HCC based on both experimental data and analyses on HCC human samples, mostly correlating the expression of Snail and Twist and the repression of E-cadherin with the poorest prognosis [41–44].
In the following sections, we review the currently known molecular mechanisms by which hepatitis B and C and Delta viruses can promote HCC progression and metastasis.
Hepatitis B virus and epithelial-mesenchymal transition
Hepatitis B virus (HBV) is a member of the Hepadnaviridae family. Ten HBV genotypes (A-J) have been described having different geographical distribution, with genotypes C and D more frequently associated with the development of HCC [45, 46].
The HBV genome is a relaxed circular and partially double-stranded DNA molecule of about 3.2 kb with four overlapping open reading frame (ORF) encoding the viral proteins S, C, P, and X. The ORF S contains three start sites which generate the synthesis of three different sized surface proteins (small, middle, and large) of the envelope (HBsAg). The ORF C encodes for both the secreted precore (HBeAg) and the core (HBcAg) proteins of the nucleocapsid [47]. The ORF P generates the viral polymerase which shows DNA polymerase, reverse transcriptase, and RNaseH activities [48]. Finally, the ORF X encodes for the HBx protein, which not only seems to be important for viral replication but is also known to modulate a number of processes in the host cell such as transcription, signal transduction, cell cycle progression, and apoptosis [49].
Replication of the HBV genome occurs within the nucleus of the infected hepatocytes, where it integrates into the host genome, thus disrupting gene functionality and making cells more susceptible to malignant transformation [47]. On the other hand, integration in the host genome could also result in truncated or mutated viral proteins which may direct oncogenesis as well [50].
Several papers report the ability of HBV to promote HCC invasiveness through induction of EMT. A major role in this process has been attributed to the viral HBx protein, which was found, since the early 2000 to disrupt cell-cell junctions and cell-extracellular matrix interactions and to promote cell migration [51, 52].
Among the mechanisms by which HBx protein can trigger EMT, there is the activation of signal transducers and activators of transcription (STAT) proteins, and specifically STAT5 [53] and STAT3 [54]. STAT proteins are a family of seven proteins (STAT1–4, 5a, 5b, and 6) activated by several cytokines and growth factors by means of tyrosine phosphorylation. After dimerization, they move to the nucleus where they serve as transcription factors [55, 56]. STAT proteins were found persistently activated in a number of cancers, including HCC [55], and have been also associated with tumor invasion and metastasis [57].
STAT5b was found activated in human samples from patients affected by aggressive HBV-associated HCC with an adverse clinical outcome. In vitro experiments on HBx-transfected hepatoma cells showed this activation to be at least in part due to HBx protein and to trigger a switch from epithelial to mesenchymal cell morphology together with the loss of E-cadherin and gain of vimentin [53]. More recently, HBx protein has been linked to the activation of STAT3, which in turn promotes EMT in cultured liver cells by upregulating the EMT-related transcription factor Twist [54].
HBx protein can trigger EMT by activating other signal transduction pathways, such as the ones mediated by c-Src and PI3K/AKT/GSK-3b respectively [58, 59]. The proto-oncogene c-Src is a non-receptor tyrosine kinase involved in the progression of a variety of tumors. When transfected into a human HCC cell line, HBx protein was found to significantly increase c-Src activity, thus increasing cell invasiveness and inducing changes in cell morphology and in the expression of epithelial and mesenchymal markers, which were reversed upon c-Src inhibition [59].
The PI3K/AKT/GSK-3b pathway, by regulating cell cycle at multiple levels, is a well-known mechanism of tumor progression [60]. It was recently demonstrated that HBx protein can activate this pathway, resulting in the stabilization of the transcription factor Snail at the protein level. This contributes to promote EMT and tumor invasion both in vitro and in vivo [58]. The same AKT/GSK-3b pathway is activated by FAT10, an oncogene belonging to the family of the ubiquitin-like modifiers, which is upregulated in HCC and other cancers [61, 62], and whose expression was found to positively correlate with invasive colon cancer [63]. Increased expression of FAT10 was observed in HBV-associated HCC patients and was shown to correlate with poor prognosis [64]. Moreover, FAT10 over-expression was able to promote EMT in vitro, as deduced by cell invasiveness, decreased expression of E-cadherin, and increased expression of both N-cadherin and vimentin [64].
Finally, HBx protein can induce EMT through epigenetic mechanisms. This viral protein was found to regulate E-cadherin expression through hypermethylation of the promoter [65], histone deacetylation of the gene, and suppression of miRNA-373, a positive regulator of E-cadherin expression [66]. Furthermore, HBx protein can affect the expression of miRNA-148a which was shown to suppress EMT in vitro and to inhibit HCC metastasis in vivo by targeting the transcription factor hematopoietic pre-B cell leukemia transcription factor-interacting protein (HPIP) and its downstream AKT/ERK/FOXO4/ATF5 pathway, culminating in mTOR regulation [67].
Besides the aforesaid molecular mechanisms of EMT induction, other less strong or less characterized correlations between HBV infection and HCC invasiveness have been reported. A potential link may exist between EMT in HBV-associated HCC and the expression of the stemness marker antigen CD133 [68], the increased expression of the polymeric immunoglobulin receptor (pIgR) [69] and the increased expression of T cell lymphoma invasion and metastasis 2 (TIAM2) gene [70].
Hepatitis C virus and epithelial-mesenchymal transition
HCV is a member of the Flaviviridae family. Six genotypes (HCV1 to HCV6) with different geographical pattern are known, each being actually further classified into subtypes [71]. It has been demonstrated that different genotypes specifically and differentially regulate host gene expression, and this probably may account for different clinical manifestations of the disease [71–74].
The HCV genome is a positive-sense, single-stranded RNA molecule of about 9.6 kb. It carries a single ORF encoding a precursor polyprotein which is cleaved into three structural (core, E1, and E2) and seven non-structural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins [75, 76] by both viral and cellular proteases.
E1 and E2 are two envelope glycoproteins determinant for HCV infection since they allow the binding of the virus to the host cell’s receptor and its endocytosis-mediated entry [77, 78].
The HCV core protein is required to form nucleocapsid and protect the viral genome. It consists of three domains with different functions. The domain I binds RNA, thus ensuring the assembly of the capsid. The domain II interacts with lipids and membrane proteins, and the domain III is essential for the stability of immature core protein but it is removed from the mature protein [79].
The p7 protein is an ion channel participating in capsid assembly and envelopment, and together with the NS2, it is required for progeny production [80]. NS2/3 is an autoprotease performing the intramolecular cleavage between NS2, probably involved in modulating host cell gene expression and apoptosis, and NS3 [81]. NS3-4A serine protease is a non-covalent dimer: NS3 has both a helicase and a serine protease domain. NS4A has a cofactor function. Together, they induce four proteolytic cleavages on the HCV polyprotein that included their self-cleavage [82]. NS4B is involved in the formation of a membranous platform in which viral replication takes place [83] . NS5A seems to have no enzymatic activity, but it takes part in several but still unclear viral and cellular processes [84]. NS5B is a RNA-dependent RNA polymerase, an essential protein for viral RNA replication [85]. NS3, NS4A, NS4B, NS5A, and NS5B proteins form a replicase complex necessary for the production of viral proteins and RNA [75].
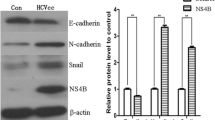
HCV replicates in the cytoplasm, and its RNA genome does not integrate into the host one. HCC, however, may arise as a consequence of complex interactions between viral and cellular proteins, affecting a number of different cell processes such as cell cycle, apoptosis, signal transduction, lipid metabolism, transcription regulation, and immune response [86, 87]. Moreover, HCV also plays a role in the later stages of hepatocarcinogenesis by promoting invasiveness and metastasis through induction of EMT. The ability of HCV to trigger EMT has been demonstrated in vitro in both infected human primary hepatocytes and human hepatoma cells [88, 89]. In Bose et al., both HCV genotypes tested, namely 1a and 2a, were similarly successful in repressing the epithelial marker E-cadherin, in upregulating mesenchymal markers (vimentin, snail, slug, twist, and nuclear β-catenin) and in inducing morphological changes in the cells [88].
Recently, Iqbal et al. identified osteopontin as a mediator of HCV-induced EMT, migration, and invasiveness in cultured hepatoma cells [89, 90]. Osteopontin (OPN) is a secreted protein interacting with integrins and CD44 cell surface receptors, which is known not only to mediate signal transduction but also to influence tumor microenvironment, thereby favoring cancer progression and invasion [91, 92]. HCV not only causes OPN activation through Ca(2+) signaling and ROS increase [90] but also stimulates its expression and secretion, thus resulting in the activation of cellular kinases such as FAK, Akt, and Src. The latter event mediates E-cadherin repression, N-cadherin upregulation, and increased cell migration and invasion [89].
In a number of papers, specifically, the HCV core protein has been reported as responsible of EMT promotion. One way by which the HCV core protein can induce EMT is by involving the TFG-β signaling. TGF-β can signal through both a canonical SMAD-dependent and other non-SMAD pathways (such as the ones mediated by MAP kinases, Wnt, NF-kB, Notch, AKT, and others). In the SMAD pathway, upon binding to its receptors, TFG-β recruits SMAD2/3, and after SMAD2 phospohorylation and SMAD4 recruitment, this complex moves to the nucleus where it mediates transcriptional regulation of target genes. In normal cells, TFG-β functions as an inhibitor of proliferation, blocking cell cycle progression by decreasing the expression of c-Myc and cyclin-dependent kinases and by increasing that of cyclin-dependent kinase inhibitors. Moreover, TFG-β is also known to induce apoptosis [93, 94]. TFG-β, however, can switch from being a tumor suppressor to being a tumor promoter especially in advanced cancer, triggering metastasis through activation of EMT [95, 96].
HCV core protein derived from HCC was found to attenuate the cytostatic and the pro-apoptotic effect of TGF-β on both mouse and human primary hepatocytes and on the human hepatoma cell line Huh7. In contrast, the core protein was able to induce EMT through TGF-β activation in the same cell systems, as demonstrated by the acquisition of a spindle-like cell morphology, the increased mesenchymal markers alpha smooth muscle actin (α-SMA) and vimentin, and the decreased epithelial marker E-cadherin. All these effects were shown to be mediated by decreased activation of Smad3 [97], which was found to physically interact with the core protein [98].
HCV core protein may also epigenetically promote EMT. Indeed, in human hepatoma cells, it was shown to repress the expression of FSRP1, a modulator of the Wnt//β-catenin pathway which is involved in several cancers as well as in EMT. The core protein supports the methylation through DNA methyltransferase-1 (Dnmt1) and the deacetylation through histone deacetylase-1(HDAC1) of the SFRP1 promoter, and this results in SFRP1 silencing, which in turn induces EMT, as proved by the downregulation of E-cadherin; the upregulation of the mesenchymal markers β-catenin, fibronectin, and twist; and by the increased migratory and invasive potential of the cells [99]. All these effects were reversed by Dnmt1 knockdown and SFRP1 overexpression, which also reduced tumor growth and aggressiveness in a in vivo model of HCC [99]. Furthermore, in vitro evidences support that the HCV core protein, in particular, belonging to the virus genotype 1b represses E-cadherin expression by hypermethylating its promoter [14, 34]. This epigenetic modification is likely achieved, thanks to the ability of the core protein to activate DNMT1 and DNMT3b expression [34, 100].
A role for HCV core protein in EMT induction was also demonstrated in cholangiocarcinoma, another type of HCV-associated liver cancer. Here, the invasiveness and the metastatic potential accompanied by E-cadherin decrease and fibronectin and vimentin increase were correlated to core-dependent activation of LOXL2 [101], which is emerging as promoter of EMT through its interaction with and stabilization of the EMT-related transcription factor Snail [102, 103].
Also, some non-structural HCV proteins, namely NS3-NS4A, and NS5A, have been found to promote EMT. Verga-Gérard et al. showed NS3A-4A to induce EMT through the enhancement of TGF-β signaling [104].
In Akkari et al., NS5A was found to induce EMT by increasing levels of the mesenchymal markers N-cadherin and vimentin through the activation of TWIST2 [105]. Moreover, NS5A strengthens the action of the oncogene RAS and of the cytokine TGF-β in disrupting cell polarity and promoting cell transformation, migration, and metastasis [105].
Hepatitis D virus and epithelial-mesenchymal transition
Hepatitis delta virus (HDV) is a defective RNA virus whose infection only occurs in individuals previously or simultaneously infected with HBV. The first case is known as co-infection, while the second as super-infection. The presence of both viruses induces a more severe disease than HBV infection alone [106, 107]. HDV is a small spherical particle discovered for the first time in 1970s by Rizzetto and colleagues [108]. The external coat of the virus contains the HBV large, medium, and small B surface antigens (HBsAg) and wraps a nucleocapsid formed by a circular single-stranded negative RNA complexed to about 200 molecules of hepatitis D antigen (HDAg) per genome. The receptor on the host cell is thought to be the same of HBV since the two viruses share their coat protein. After entering in the hepatocyte, the viral complex RNA-HDAg translocates into the nucleus. HDV is not equipped with a own RNA polymerase that is why it needs the RNA polymerase of the host cell [107].
Only two isoforms of a unique protein are known to be translated from the HDV genome, namely the large-HDAg (L-HDAg) and the small-HDAg (S-HDAg). These isoforms only differ in length by 19 amino acids and are produced by RNA editing: when a stop codon UAG in the S-HDAg is changed in UGG through adenosine deamination, the L-HDAg is generated. The S-HDAg is necessary for viral replication; on the contrary, the L-HDAg, although required for virus assembly, seems to inhibit RNA replication [107, 109].
Eight HDV genotypes with different distribution in the world have been identified [107, 110]. Moreover, due to the high evolutionary rate of HDV genome, each genotype can be further classified in quasispecies co-existing in the same patient [107]. A recent study has demonstrated that certain quasispecies with high replicative potential are more capable of inducing EMT by increasing the expression of mesenchymal markers such as vimentin, Twist, and Snail and by decreasing the expression of the epithelial cell-cell adhesion molecule E-cadherin, as observed when used to transfect hepatoma cells.
In more detail, the L-HDAg was shown to be more involved than the S-HDAg in EMT promotion since cells transfected with the L-HDAg exhibit significative alteration of the above cited EMT markers while cells transfected with the S-HDAg do not [111]. Moreover, TGF-β, another factor implicated in liver fibrosis and cirrhosis [112] and in EMT [28], results to be more produced in the culture medium of cells transfected with L-HDAg than in cells transfected with S-HDAg. These in vitro data are supported by clinical observation that HDV quasispecies with lower EMT-inducing activity are associated with disease remission [111]. Moreover, since HDV, like HCV, was shown to induce DNMT3b expression in Huh-7 liver cancer cells [113], one might speculate that also HDV may promote EMT epigenetically.
Conclusions
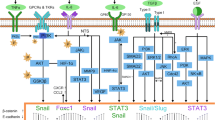
Hepatitis viruses, especially HBV and HCV, are the main causative agents of hepatocellular carcinoma onset, and more recently, they have been also recognized as mediators of cancer progression. Indeed, several evidences both at the experimental and clinical level show that hepatitis viruses are capable of promoting epithelial-mesenchymal transition and metastasis through the activation of different pathways (Fig. 1).

Scheme illustrating the EMT induction by hepatitis viruses HBV, HCV, and HDV through different molecular pathways. The resulting loss of cell polarity and intercellular junctions enables tumor cells to cross extracellular matrix to spread and metastasize
Currently, the most effective approaches to HCC treatment are represented by surgical resection, liver transplantation, and local ablation even though their application is limited to patients with early stage tumors. As for advanced stage HCC, instead, therapies are mainly palliative or symptomatic, except for the use of sorafenib, whose efficacy not only relies on its anti-angiogenetic activity but also on its newly discovered ability to interfere with EMT at different levels [114–117]. Sorafenib, in fact, was reported to suppress TGF-β signaling, activated in EMT, by blocking the downstream phosphorylation of Smad2/3 and STAT3 [114, 117]. Moreover, it was found to epigenetically regulate EMT-associated genes through reversing the state of histone acetylation and methylation on the promoters of TGF-β1, Snail, and snail homolog-2 (Slug) and through inhibiting the switch from E-cadherin to N-cadherin [117].
It should be noted, however, that the response to Sorafenib is low [24], and acquired resistance to this drug is often observed [25].
A better understanding of the molecular bases of cancer progression should be useful to develop new therapeutic approaches for the treatment of advanced disease. Studying in more detail and directly targeting the pathways involved in virus-induced EMT should be useful for blocking or slowing the progression of hepatitis viruses-related HCC.
References
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27.
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–55.
Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300–8.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87.
Bartosch B. Hepatitis B and C viruses and hepatocellular carcinoma. Viruses. 2010;2:1504–9.
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–73. e1261.
Abbas Z, Qureshi M, Hamid S, Jafri W. Hepatocellular carcinoma in hepatitis D: does it differ from hepatitis B monoinfection? Saudi J Gastroenterol. 2012;18:18–22.
Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:790–2.
Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, de Franchis R, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136:1629–38.
Verme G, Brunetto MR, Oliveri F, Baldi M, Forzani B, Piantino P, et al. Role of hepatitis delta virus infection in hepatocellular carcinoma. Dig Dis Sci. 1991;36:1134–6.
Bouchard MJ, Navas-Martin S. Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett. 2011;305:123–43.
Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834–47.
Ripoli M, Barbano R, Balsamo T, Piccoli C, Brunetti V, Coco M, et al. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res. 2011;160:74–81.
Rongrui L, Na H, Zongfang L, Fanpu J, Shiwen J: Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr Pharm Des 2013.
Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093–107.
Shimoda R, Nagashima M, Sakamoto M, Yamaguchi N, Hirohashi S, Yokota J, et al. Increased formation of oxidative DNA damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 1994;54:3171–2.
Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, et al. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1994;91:12808–12.
Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–7.
Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–70.
Lin S, Hoffmann K, Schemmer P. Treatment of Hepatocellular Carcinoma: A Systematic Review. Liver Cancer. 2012;1:144–58.
Vivarelli M, Montalti R, Risaliti A. Multimodal treatment of hepatocellular carcinoma on cirrhosis: an update. World J Gastroenterol. 2013;19:7316–26.
Xie B, Wang DH, Spechler SJ. Sorafenib for treatment of hepatocellular carcinoma: a systematic review. Dig Dis Sci. 2012;57:1122–9.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol Res. 2013;43:147–54.
Lee HS. Management of patients with hepatocellular carcinoma and extrahepatic metastasis. Dig Dis. 2011;29:333–8.
Wang C, Jiang K, Kang X, Gao D, Sun C, Li Y, et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol. 2012;44:2308–20.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Firrincieli D, Boissan M, Chignard N. Epithelial-mesenchymal transition in the liver. Gastroenterol Clin Biol. 2010;34:523–8.
Lamouille S, Derynck R. Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs. 2010;193:8–22.
van Zijl F, Zulehner G, Petz M, Schneller D, Kornauth C, Hau M, et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Futur Oncol. 2009;5:1169–79.
Yang G, Yuan J, Li K. EMT transcription factors: implication in osteosarcoma. Med Oncol. 2013;30:697.
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–37.
Arora P, Kim EO, Jung JK, Jang KL. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008;261:244–52.
Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25:593–600.
Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer Biol. 2000;10:185–200.
Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66.
Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25:695–705.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8.
Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9:1608–20.
Mima K, Hayashi H, Kuroki H, Nakagawa S, Okabe H, Chikamoto A, et al. Epithelial-mesenchymal transition expression profiles as a prognostic factor for disease-free survival in hepatocellular carcinoma: clinical significance of transforming growth factor-beta signaling. Oncol Lett. 2013;5:149–54.
Miyoshi A, Kitajima Y, Kido S, Shimonishi T, Matsuyama S, Kitahara K, et al. Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer. 2005;92:252–8.
Sugimachi K, Tanaka S, Kameyama T, Taguchi K, Aishima S, Shimada M, et al. Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin Cancer Res. 2003;9:2657–64.
Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, et al. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009;50:1464–74.
Cao GW. Clinical relevance and public health significance of hepatitis B virus genomic variations. World J Gastroenterol. 2009;15:5761–9.
Shi YH. Correlation between hepatitis B virus genotypes and clinical outcomes. Jpn J Infect Dis. 2012;65:476–82.
Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. Biomed Res Int. 2013;2013:810461.
Wei Y, Neuveut C, Tiollais P, Buendia MA. Molecular biology of the hepatitis B virus and role of the X gene. Pathol Biol (Paris). 2010;58:267–72.
Ng SA, Lee C. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol. 2011;46:974–90.
Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–61.
Lara-Pezzi E, Majano PL, Yanez-Mo M, Gomez-Gonzalo M, Carretero M, Moreno-Otero R, et al. Effect of the hepatitis B virus HBx protein on integrin-mediated adhesion to and migration on extracellular matrix. J Hepatol. 2001;34:409–15.
Lara-Pezzi E, Roche S, Andrisani OM, Sanchez-Madrid F, Lopez-Cabrera M. The hepatitis B virus HBx protein induces adherens junction disruption in a src-dependent manner. Oncogene. 2001;20:3323–31.
Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng IO, et al. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res. 2006;66:9948–56.
Teng J, Wang X, Xu Z, Tang N. HBx-dependent activation of Twist mediates STAT3 control of epithelium-mesenchymal transition of liver cells. J Cell Biochem. 2013;114:1097–104.
Gao B, Wang H, Lafdil F, Feng D. STAT proteins—key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol. 2012;57:430–41.
Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105.
Haura EB, Turkson J, Jove R. Mechanisms of disease: insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–24.
Liu H, Xu L, He H, Zhu Y, Liu J, Wang S, et al. Hepatitis B virus X protein promotes hepatoma cell invasion and metastasis by stabilizing Snail protein. Cancer Sci. 2012;103:2072–81.
Yang SZ, Zhang LD, Zhang Y, Xiong Y, Zhang YJ, Li HL, et al. HBx protein induces EMT through c-Src activation in SMMC-7721 hepatoma cell line. Biochem Biophys Res Commun. 2009;382:555–60.
Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–45.
Lee CG, Ren J, Cheong IS, Ban KH, Ooi LL. Yong Tan S, Kan A, Nuchprayoon I, Jin R, Lee KH, Choti M, Lee LA: Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene. 2003;22:2592–603.
Lukasiak S, Schiller C, Oehlschlaeger P, Schmidtke G, Krause P, Legler DF, et al. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 2008;27:6068–74.
Qing X, French BA, Oliva J, French SW. Increased expression of FAT10 in colon benign, premalignant and malignant epithelial neoplasms. Exp Mol Pathol. 2011;90:51–4.
Liu L, Dong Z, Liang J, Cao C, Sun J, Ding Y, Wu D: As an independent prognostic factor, FAT10 promotes hepatitis B virus-related hepatocellular carcinoma progression via Akt/GSK3beta pathway. Oncogene 2013
Liu J, Lian Z, Han S, Waye MM, Wang H, Wu MC, et al. Downregulation of E-cadherin by hepatitis B virus X antigen in hepatocellular carcinoma. Oncogene. 2006;25:1008–17.
Arzumanyan A, Friedman T, Kotei E, Ng IO, Lian Z, Feitelson MA. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31:563–72.
Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123:630–45.
Na DC, Lee JE, Yoo JE, Oh BK, Choi GH, Park YN. Invasion and EMT-associated genes are up-regulated in B viral hepatocellular carcinoma with high expression of CD133-human and cell culture study. Exp Mol Pathol. 2011;90:66–73.
Ai J, Tang Q, Wu Y, Xu Y, Feng T, Zhou R, et al. The role of polymeric immunoglobulin receptor in inflammation-induced tumor metastasis of human hepatocellular carcinoma. J Natl Cancer Inst. 2011;103:1696–712.
Chen JS, Su IJ, Leu YW, Young KC, Sun HS. Expression of T-cell lymphoma invasion and metastasis 2 (TIAM2) promotes proliferation and invasion of liver cancer. Int J Cancer. 2012;130:1302–13.
Ripoli M, Pazienza V. Impact of HCV genetic differences on pathobiology of disease. Expert Rev Anti Infect Ther. 2011;9:747–59.
Abid K, Pazienza V, de Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, et al. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol. 2005;42:744–51.
Pazienza V, Clement S, Pugnale P, Conzelman S, Foti M, Mangia A, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45:1164–71.
Pazienza V, Clement S, Pugnale P, Conzelman S, Pascarella S, Mangia A, et al. Gene expression profile of Huh-7 cells expressing hepatitis C virus genotype 1b or 3a core proteins. Liver Int. 2009;29:661–9.
Bartenschlager R, Lohmann V. Replication of the hepatitis C virus. Baillieres Best Pract Res Clin Gastroenterol. 2000;14:241–54.
Shrivastava S, Mukherjee A, Ray RB. Hepatitis C virus infection, microRNA and liver disease progression. World J Hepatol. 2013;5:479–86.
Fraser J, Boo I, Poumbourios P, Drummer HE. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J Biol Chem. 2011;286:31984–92.
Lavie M, Goffard A, Dubuisson J: HCV Glycoproteins: assembly of a functional E1-E2 heterodimer. 2006
Polyak SJ, Klein KC, Shoji I, Miyamura T, Lingappa JR: Assemble and interact: pleiotropic functions of the HCV core protein. 2006
Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, et al. Hepatitis c Virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 2013;9:e1003355.
Welbourn S, Pause A: HCV NS2/3 protease. 2006
Lin C: HCV NS3-4A Serine protease. 2006
Sklan EH, Glenn JS: HCV NS4B: from obscurity to central stage. 2006
Schmitz U, Tan SL. NS5A—from obscurity to new target for HCV therapy. Recent Pat Antiinfect Drug Discov. 2008;3:77–92.
Ranjith-Kumar CT, Kao CC: Biochemical activities of the HCV NS5B RNA-dependent RNA polymerase. 2006
Jeong SW, Jang JY, Chung RT. Hepatitis C virus and hepatocarcinogenesis. Clin Mol Hepatol. 2012;18:347–56.
McGivern DR, Lemon SM. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene. 2011;30:1969–83.
Bose SK, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Hepatitis C virus induces epithelial-mesenchymal transition in primary human hepatocytes. J Virol. 2012;86:13621–8.
Iqbal J, McRae S, Mai T, Banaudha K, Sarkar-Dutta M, Waris G. Role of hepatitis C virus induced osteopontin in epithelial to mesenchymal transition, migration and invasion of hepatocytes. PLoS One. 2014;9:e87464.
Iqbal J, McRae S, Banaudha K, Mai T, Waris G. Mechanism of hepatitis C virus (HCV)-induced osteopontin and its role in epithelial to mesenchymal transition of hepatocytes. J Biol Chem. 2013;288:36994–7009.
Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med. 2010;14:2037–44.
Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16:79–87.
Dooley S, ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012;347:245–56.
Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-beta—an excellent servant but a bad master. J Transl Med. 2012;10:183.
Akhurst RJ, Derynck R. TGF-beta signaling in cancer—a double-edged sword. Trends Cell Biol. 2001;11:S44–51.
Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586:1959–70.
Battaglia S, Benzoubir N, Nobilet S, Charneau P, Samuel D, Zignego AL, et al. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS One. 2009;4:e4355.
Pavio N, Battaglia S, Boucreux D, Arnulf B, Sobesky R, Hermine O, et al. Hepatitis C virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with Smad3 and inhibit the TGF-beta pathway. Oncogene. 2005;24:6119–32.
Quan H, Zhou F, Nie D, Chen Q, Cai X, Shan X, Zhou Z, Chen K, Huang A, Li S, Tang N: Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene 2013
Benegiamo G, Vinciguerra M, Mazzoccoli G, Piepoli A, Andriulli A, Pazienza V. DNA methyltransferases 1 and 3b expression in Huh-7 cells expressing HCV core protein of different genotypes. Dig Dis Sci. 2012;57:1598–603.
Li T, Li D, Cheng L, Wu H, Gao Z, Liu Z, et al. Epithelial-mesenchymal transition induced by hepatitis C virus core protein in cholangiocarcinoma. Ann Surg Oncol. 2010;17:1937–44.
Moon HJ, Finney J, Xu L, Moore D, Welch DR, Mure M. MCF-7 cells expressing nuclear associated lysyl oxidase-like 2 (LOXL2) exhibit an epithelial-to-mesenchymal transition (EMT) phenotype and are highly invasive in vitro. J Biol Chem. 2013;288:30000–8.
Peinado H, Del Carmen Iglesias-de la Cruz M, Olmeda D, Csiszar K, Fong KS, Vega S, et al. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. Embo J. 2005;24:3446–58.
Verga-Gerard A, Porcherot M, Meyniel-Schicklin L, Andre P, Lotteau V, Perrin-Cocon L. Hepatitis C virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF-beta signaling. Faseb J. 2013;27:4027–40.
Akkari L, Gregoire D, Floc'h N, Moreau M, Hernandez C, Simonin Y, et al. Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J Hepatol. 2012;57:1021–8.
Alves C, Branco C, Cunha C. Hepatitis delta virus: a peculiar virus. Adv Virol. 2013;2013:560105.
Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378:73–85.
Rizzetto M, Canese MG, Arico S, Crivelli O, Trepo C, Bonino F, et al. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18:997–1003.
Tseng CH, Lai MM. Hepatitis delta virus RNA replication. Viruses. 2009;1:818–31.
Le Gal F, Gault E, Ripault MP, Serpaggi J, Trinchet JC, Gordien E, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis. 2006;12:1447–50.
Shih HH, Sheen IJ, Su CW, Peng WL, Lin LH, Wu JC. Hepatitis D virus isolates with low replication and epithelial-mesenchymal transition-inducing activity are associated with disease remission. J Virol. 2011;86:9044–54.
Choi SH, Jeong SH, Hwang SB. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology. 2007;132:343–57.
Benegiamo G, Vinciguerra M, Guarnieri V, Niro GA, Andriulli A, Pazienza V. Hepatitis delta virus induces specific DNA methylation processes in Huh-7 liver cancer cells. FEBS Lett. 2013;587:1424–8.
Chen YL, Lv J, Ye XL, Sun MY, Xu Q, Liu CH, et al. Sorafenib inhibits transforming growth factor beta1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology. 2011;53:1708–18.
Nagai T, Arao T, Furuta K, Sakai K, Kudo K, Kaneda H, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther. 2011;10:169–77.
Smolle E, Taucher V, Petru E, Haybaeck J. Targeted treatment of ovarian cancer—the multiple-kinase-inhibitor sorafenib as a potential option. Anticancer Res. 2014;34:1519–30.
Zhang J, Chen YL, Ji G, Fang W, Gao Z, Liu Y, et al. Sorafenib inhibits epithelial-mesenchymal transition through an epigenetic-based mechanism in human lung epithelial cells. PLoS One. 2013;8:e64954.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Panebianco, C., Saracino, C. & Pazienza, V. Epithelial-mesenchymal transition: molecular pathways of hepatitis viruses-induced hepatocellular carcinoma progression. Tumor Biol. 35, 7307–7315 (2014). https://doi.org/10.1007/s13277-014-2075-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2075-x