
Abstract
Chronic myelogenous leukemia (CML) is a complex disease with a genetic basis. The genetic association studies (GASs) that have investigated the association between adult CML and 5,10-methylenetetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphisms have produced contradictory and inconclusive results. The aim of this meta-analysis is to provide a relatively comprehensive assessment of the association of these polymorphisms with adult CML risk. A literature search for eligible GAS published before September 15, 2013 was conducted in PubMed, Embase, Web of Science, Cochrane Library, and China National Knowledge Infrastructure (CNKI) databases. Pooled odds ratios (ORs) with their corresponding 95 % confidence intervals (95 % CIs) were used to evaluate the strength of the association under a fixed or random effect model according to heterogeneity test results. All analyses were performed using the Stata software, version 12.0. Twelve case-control studies were included in this meta-analysis with a total of 932 CML patients and 3,465 healthy controls. For MTHFR C677T (dbSNP: rs1801133, C>T), though the pooled ORs were not significant in the overall population, all the ORs greater than 1 suggested an increased risk of CML for carriers of the risk allele. However, stratified analysis based on genotyping method revealed a significant association in the PCR-restriction fragment length polymorphism (RFLP) subgroup, possibly as a result of heterogeneity. For MTHFR A1298C (dbSNP: rs1801131, A>C), the combined results showed that carriers of the C allele may be associated with a decreased risk of adult CML. Stratified analysis showed that the magnitude of this effect was especially significant among Asians, indicating ethnicity differences in adult CML susceptibility. This meta-analysis shows that the C allele of MTHFR A1298C may be associated with a decreased risk in adult CML, especially among Asians, while MTHFR C677T may not be associated with adult CML risk. However, the development of adult CML may be the result of gene-gene and gene-environment interactions, which should be considered in future individual GAS and subsequent meta-analyses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leukemias, as a group, are cancers that develop from hematopoietic cell lines [1]. Genetic translocations, inversions, or deletions in hematopoietic cells disrupt the normal function of the genes at these locations, altering normal blood cell development [2]. As a result, dysfunctional or non-differentiated leukemic cells accumulate in the bone marrow and progressively replace normal hematopoietic cells [3]. Leukemias can be either acute or chronic and can arise from myeloid or lymphoid cell lines [4, 5]. Chronic myelogenous leukemia (CML), one of the major forms of leukemias, is a clonal, myeloproliferative disease characterized by the accumulation of myeloid precursors in the bone marrow, blood, and body tissues [6]. It is a relatively rare disease worldwide, accounting for approximately 14 % of all leukemias [7]. Although the clinical and biological aspects of CML are well documented, little is known about the factors that condition an individual’s susceptibility to CML. Since leukemias are derived from rapidly proliferating tissues that have the greatest requirement for DNA synthesis, it is thought that CML are likely to be affected by the metabolic fate of folic acid [8]. Folic acid metabolism plays an essential role in DNA synthesis and methylation processes, which are associated with CML, and 5,10-methylenetetrahydrofolate reductase (MTHFR) is a central component of the human folic acid metabolic pathway as shown in Fig. 1 [9]. Folate deficiency has been associated with hypomethylation and uracil misincorporation into DNA during replication, increasing the risk of chromosomal aberrations and presumably facilitating the onset of oncogenic processes [10]. Folate metabolism requires the optimal activity of various enzymes [11]. MTHFR is one of the most important enzymes of the folate cycle and affects both nucleotide synthesis and methylation reactions [12]. It catalyzes the reduction of 5,10-methylenetetrahydrofolate (5,10-methyleneTHF) to 5,10-methyltetrahydrofolate (5,10-methylTHF), which is the predominant circulatory form of folate and carbon donor for the remethylation of homocysteine to methionine [13]. The enhanced availability of 5,10-methyleneTHF may play a protective role in the onset of CML by reducing the misincorporation of uracil into DNA, which might otherwise result in double-strand breaks during uracil excision repair processes [14, 15]. This protective effect might be due to more efficient DNA synthesis and repair, since excessive 5,10-methyleneTHF levels are used in the conversion of uracil to thymine and for purine synthesis.

Overview of the human folic acid metabolic pathway and the role of MTHFR
The MTHFR gene, which encodes MTHFR enzyme, is located on the short arm of chromosome 1 at position 36.3 [16]. Two common polymorphisms have been described in the MTHFR gene which are single nucleotide substitutions resulting in amino acid changes: (1) C677T (exon 4 at codon 222), which is a C to T substitution at position 677 resulting in an alanine to valine substitution, and (2) A1298C (exon 7 at codon 429), which causes a glutamate to alanine (A to C) substitution [17]. Both of these polymorphisms have been found to result in decreased MTHFR enzyme activity, leading to increased homocysteine levels and, thus, to an imbalance in plasma folate concentration [18]. To date, several genetic association studies (GASs) have been carried out to investigate the role of MTHFR C677T and A1298C in the development of CML among various populations. However, these studies have produced contradicted and inconclusive results, partly because they had limited sample sizes and insufficient statistical power to demonstrate a significant association. In addition, these studies included different populations and sampling strategies, making their results difficult to interpret. Hence, we performed a meta-analysis to derive a relatively comprehensive assessment of the relationship between the MTHFR C677T and A1298C polymorphisms and the risk of adult CML. To evaluate this association, we analyzed the pooled data from all available GAS relating the MTHFR C677T and A1298C variants with adult CML risk.
Materials and methods
Identification and eligibility of relevant studies
A literature search for GAS that investigated the association between the MTHFR C677T or A1298C genetic variants and the risk of adult CML published before September 15, 2013 was conducted in the following electronic databases: PubMed, Embase, Web of Science, Cochrane Library, and China National Knowledge Infrastructure (CNKI) databases. The following combined MeSH terms were used: (“methylenetetrahydrofolate reductase” or “MTHFR”) and (“chronic myelogenous leukemia” or “CML”) and (“genetic polymorphism” or “SNP”). The search was done without limitations on language but only included those studies that were conducted on human subjects. All references in eligible articles were extensively reviewed to identify additional published articles.
To be included in the analysis, candidate studies had to meet the following criteria: (1) case-control study focused on the relationship between MTHFR C677T or A1298C and adult CML risk, (2) all patients who met the diagnostic criteria for CML, and (3) included sufficient original data for calculating odds ratios (ORs) with corresponding 95 % confidence intervals (95 % CIs). The major reasons for excluding studies were as follows: (1) not case-control study, (2) duplicate publications, and (3) no available data reported. For multiple studies using overlapping cases or controls, the study with the largest sample size was included in the meta-analysis. This meta-analysis was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidance with slight modification to be more suitable for this study and did not require ethics board approval [19].
Data extraction
According to the PRISMA guidance, two independent reviewers checked each full-text report for eligibility and extracted and tabulated the following data from eligible studies: surname of first author, year of publication, country of origin, ethnicity, definition and numbers of cases and controls, age, sex ratio, genotyping method, allele and genotype frequency, and Hardy-Weinberg equilibrium (HWE) status in controls. Disagreements were solved by discussion between all authors until consensus was reached. For data not provided in table form or in the main text, required information was obtained by contacting corresponding authors when possible.
Quality assessment
Strengthening the reporting of genetic association studies (STREGA) quality score system and the Newcastle-Ottawa Scale (NOS) criteria were used to assess the qualities of all included studies [20, 21]. The STREGA system includes 22 assessment items related to quality appraisal with scores ranging from 0 to 22. The included studies were classified into three levels based on their scores: low quality (0–12), moderate to high quality (13–17), and high quality (18–22). The NOS criteria use a “star” rating system to judge methodological quality based on three aspects of a study: selection, comparability, and exposure. Scores range from 0 star (worst) to 9 stars (best), with scores of 5 or higher indicating a moderate to high methodological quality. Two authors independently assessed the quality of included studies. Discrepancies over quality scores were solved by discussion with all authors and subsequent consensus.
Statistical analysis
Taking into consideration the possible between-study heterogeneity, a statistical test for heterogeneity was conducted using Cochran’s Q statistic [22]. Heterogeneity was also assessed with the I 2 metric, which calculates values between 0 and 100 % with higher values denoting a greater degree of heterogeneity [23]. When no heterogeneity was found with P > 0.05 or I 2 < 50 %, a fixed effect model was used to estimate the pooled ORs with their corresponding 95 % CIs under five genetic models: the allele model, the dominant model, the recessive model, the homozygous model, and the heterozygous model. Otherwise, a random effects model was applied. The significance of the pooled ORs was determined using the Z test. Genotype distributions in the controls were tested for conforming to HWE using the chi-square test. Subgroup analyses were performed to explain heterogeneity and to investigate whether overall reported associations were present in subgroups based on ethnicity (Caucasians, Asians, and mixed) or genotyping method (PCR-restriction fragment length polymorphism (RFLP) and non-PCR-RFLP), where applicable. Univariate and multivariate meta-regression analyses were also performed to identify variables that could possibly explain sources of heterogeneity [24]. Sensitivity analyses were conducted by omitting individual studies in turn to reflect the influence of individual datasets on the pooled results [25]. Begg’s funnel plot and Egger’s linear regression test were used to assess the potential for publication bias [26, 27]. All two-tailed P < 0.05 were considered statistically significant. All analyses were performed using the Stata software, version 12.0 (Stata Corp, College Station, TX, USA).
Results
Baseline characteristics of included studies
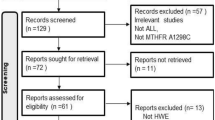
A total of 169 relevant papers were identified using the pre-specified search strategy. In accordance with the inclusion criteria, 12 case-control studies [28–38], with 12 on MTHFR C677T and 8 on MTHFR A1298C, were included in this meta-analysis. Figure 2 presents a flow chart of retrieved and excluded studies with their reasons for exclusion. A total of 4,397 subjects were involved in this meta-analysis, including 932 CML patients and 3,465 healthy controls. Studies were conducted in various populations of different ethnicities, with six on Asian, five on Caucasian, and one on mixed populations. The publication years of included studies ranged from 2003 to 2012. The distribution of genotypes in the controls was consistent with HWE in all studies (all P > 0.05). The quality scores of all the included studies were moderate to high, with STREGA scores higher than 13 and NOS stars more than 5. The characteristics and methodological quality of the included studies are summarized in Table 1.

Flow diagram of the selection of studies and specific reasons for exclusion from the present meta-analysis
Association between the MTHFR C677T polymorphism and adult CML risk
An evaluation of the association between the MTHFR C677T (dbSNP: rs1801133, C > T) polymorphism and adult CML risk is summarized in Table 2. Twelve case-control studies investigated the relationship between C677T and adult CML risk with a total of 932 CML patients and 3,465 healthy controls. The overall analysis of the association between the 677T allele and the risk of CML compared to the 677C allele revealed a nonsignificant association (P = 0.464) and significant heterogeneity (P = 0.035) under the allele model, indicating a lack of sufficient evidence for this association. In a subgroup analysis based on ethnicity, no significant results were observed for the Asian and Caucasian subgroups under the major genetic models (Fig. 3a). A marginally significant association was shown for mixed populations under the homozygous model (TT vs. CC: OR = 3.73, 95 % CI 1.02–13.68, P = 0.047), but this result may lack a statistical power due to a small sample size since only one study was included in this subgroup. A stratified analysis based on genotyping method revealed a significant association in the PCR-RFLP subgroup (T allele vs. C allele: OR = 1.23, 95 % CI 1.03–1.47, P = 0.025; TT vs. CC + CT: OR = 1.68, 95 % CI 1.12–2.50, P = 0.011; TT vs. CC: OR = 1.74, 95 % CI 1.15–2.64, P = 0.009; TT vs. CT: OR = 1.61, 95 % CI 1.05–2.46, P = 0.027) (Fig. 3b), which may be a result of heterogeneity.

Forest plots of ORs for the association between the MTHFR C677T polymorphism and susceptibility to adult CML in subgroup analysis based on ethnicity (a) and genotyping method (b) under the allele model
Association between the MTHFR A1298C polymorphism and adult CML risk
A summary of findings on the relationship between the MTHFR A1298C (dbSNP: rs1801131, A>C) polymorphism and susceptibility to adult CML is also provided in Table 2. Data from eight case-control studies, in total, comprised of 663 adult CML cases and 2,974 healthy controls, were pooled together for analysis. The fixed effects model was conducted since no heterogeneity obviously existed (all P > 0.05 and I 2 < 50 % under five genetic models). The combined results showed that carriers of the C allele may be associated with a decreased risk of CML (C allele vs. A allele: OR = 0.80, 95 % CI 0.69–0.93, P = 0.003; AC + CC vs. AA: OR = 0.79, 95 % CI 0.65–0.95, P = 0.011; CC vs. AA + AC: OR = 0.64, 95 % CI 0.44–0.94, P = 0.023; CC vs. AA: OR = 0.60, 95 % CI 0.40–0.89, P = 0.012). Stratified analysis showed that the magnitude of the effect was especially significant among Asians (C allele vs. A allele: OR = 0.80, 95 % CI 0.69–0.93, P = 0.003; AC + CC vs. AA: OR = 0.79, 95 % CI 0.65–0.95, P = 0.011; CC vs. AA + AC: OR = 0.64, 95 % CI 0.44–0.94, P = 0.023; CC vs. AA: OR = 0.60, 95 % CI 0.40–0.89, P = 0.012) (Fig. 4a), indicating ethnicity differences for adult CML risk. A significant association was also detected in the non-PCR-RFLP subgroup under two major genetic models (C allele vs. A allele: OR = 0.76, 95 % CI 0.62–0.95, P = 0.015; AC + CC vs. AA: OR = 0.75, 95 % CI 0.58–0.97, P = 0.026) (Fig. 4b).

Forest plots of ORs for the association between the MTHFR A1298C polymorphism and susceptibility to adult CML in subgroup analysis based on ethnicity (a) and genotyping method (b) under the allele model
Univariate and multivariate meta-regressions
Potential sources of between-study heterogeneity were also investigated using univariate and multivariate meta-regressions. Table 3 shows the influence of publication year, ethnicity, genotyping method, and quality score on the between-study effect size in meta-regression. For MTHFR C677T, results of meta-regression analysis revealed that publication year (P = 0.756), ethnicity (P = 0.267), STREGA score (P = 0.463), and NOS star (P = 0.760) did not significantly explain between-study heterogeneity under multivariate regression. By contrast, genotyping method (P = 0.033) was significantly correlated with the magnitude of the genetic effect, explaining more than 16 % of the heterogeneity. Regarding the variant A1298C, ethnicity was indicated as the major source of between-study heterogeneity (P = 0.032), which confirmed the ethnicity difference for adult CML susceptibility.
Sensitivity analyses and publication bias
Sensitivity analyses of both MTHFR C677T and A1298C indicated that no single study significantly influenced the pooled ORs, suggesting that the results of this meta-analysis are stable (Fig. 5). Begg’s funnel plots and Egger’s linear regression test were used to assess the potential publication bias of included studies under the allele model. The shapes of the funnel plots did not reveal any evidence of obvious asymmetry (Fig. 6). In addition, we did not find any evidence of publication bias from Egger’s linear regression test (MTHFR C677T: t = −0.16, P = 0.879; MTHFR A1298C: t = −0.25, P = 0.809). The above tests indicated a promising level of robustness and accuracy for the results of this meta-analysis.

Sensitivity analysis of the association between the MTHFR C677T (a) and MTHFR A1298C (b) polymorphisms and susceptibility to adult CML under the allele model

Begg’s funnel plots of publication bias for the association between the MTHFR C677T (a) and MTHFR A1298C (b) polymorphisms and susceptibility to adult CML under the allele model
Discussion
The role of folate in cancer is probably due to defects in different, but related, branches of folate metabolism, including the defective cell division caused by a shortage of thymidine for DNA synthesis and a shortage of methyl groups for DNA methylation [39, 40]. Folic acid metabolism plays an important role in CML, and the MTHFR gene is central in this process (Fig. 1). Common polymorphisms in the MTHFR gene have been identified as resulting in reduced MTHFR activity that decreases the pool of 5,10-methylTHF and increases the pool of 5,10-methyleneTHF. On the one hand, impaired MTHFR activity, because of polymorphic variation, reduces the amount of 5,10-methylTHF available for the methylation of homocysteine to methionine. On the other hand, the enhanced availability of 5,10-methyleneTHF may play a protective role in the onset of CML by reducing the misincorporation of uracil into DNA, which might otherwise result in double-strand breaks during uracil excision repair processes [14, 15].
The MTHFR C677T and A1298C polymorphisms have been the focus of many investigations of genetic variation in the folate metabolic pathway. However, results are inconsistent, with some studies reporting protective effects for 677T allele and 1298C allele [30, 31, 36] and others, yielding a lack of or contrary evidence of this effect [32, 37]. Hur et al. first reported a significant association between the A1298C polymorphism in MTHFR and a decreased risk of CML in the Korean population [30], and Hussain et al. found similar effects for C677T on adult CML risk in the North Indian population [31]. In addition, Lordelo et al. conducted a Brazilian case-control study analyzing the connection between the MTHFR C677T and A1298C polymorphisms and CML risk [36]. They found the two loci to be associated with CML. However, other studies have obtained conflicting results. Ismail et al. and Moon et al. studied the association between the different genotypes of MTHFR C677T and A1298C and the risk of CML [32, 37]. Both of their results showed an increased risk of CML for carriers of 677T and 1298C alleles. Other GAS failed to find strong evidence for the influence of these MTHFR variants on adult CML [28, 29, 33–35, 38]. There are several possible reasons for these inconsistencies, one of which relates to the small case population of most previous studies. In addition, it is probable that the complexity of the folate metabolic pathway may be important as MTHFR is only one of more than 30 enzymes involved in the pathway.
Meta-analysis has the advantage of synthesizing data from published GAS to provide a greater statistical power for detecting significant associations than an individual GAS, especially in the absence of large heterogeneity between studies [41]. A large number of meta-analyses have been conducted to investigate the association between the MTHFR gene and various kind of carcinomas, including prostate cancer [42, 43], lung cancer [44], gastric cancer [45, 46], breast cancer [47], ovarian cancer [48], cervical cancer [49, 50], and colorectal cancer [51]. In addition, several recent meta-analyses have investigated the association between the MTHFR gene and acute lymphoblastic leukemia (ALL), most of which concluded that both adults and children with the variant forms of MTHFR C677T and A1298C have a decreased risk of acute lymphoid leukemias [52–55]. To the best of our knowledge, our study is the first meta-analysis to describe the associations of the MTHFR genetic polymorphisms with adult CML risk. This systematic review provides a more comprehensive summary of the currently available evidence on the associations between the MTHFR C677T and A1298C polymorphisms and the risk of adult CML. In this meta-analysis, MTHFR A1298C was found to be associated with decreased adult CML susceptibility in the overall population, whereas the association between MTHFR C677T and adult CML risk was not significant. The lack of significant association between the MTHFR C677T variants and adult CML may be due to other unidentified functional variants that exist in the folate pathway and the MTHFR gene and thus influence susceptibility to CML.
In a meta-analysis, heterogeneity evaluation is always conducted. Thus, subgroup analyses based on ethnicity and genotyping method, as well as meta-regressions, were applied to find potential sources of between-study heterogeneity. In the stratified analysis by ethnicity, significant associations were found in Asian populations for MTHFR A1298C. However, few significant results were detected for MTHFR C677T. The subgroup analysis based on genotyping method indicated a significant association in the PCR-RFLP subgroup for MTHFR C677T, but not in the PCR-RFLP subgroup for MTHFR A1298C. There are several possible reasons for such differences. First, the distribution of the risk allele or genotype may vary between ethnicities. In addition, clinical heterogeneity such as age, sex ratio, BMI, years from onset, and disease severity may also explain discrepancies. Moreover, such different results could also be explained by study design or sample size. Results of meta-regression analyses further confirmed the sources of heterogeneity for both variants, revealing that genotyping method could explain more than 16 % of the heterogeneity for MTHFR C677T, and ethnicity might be the major source of between-study heterogeneity.
In interpreting the results of this meta-analysis, some specific issues need to be addressed. First, only published studies were included, and as a result, publication bias may have occurred, though not shown by statistical analysis. Second, as with other complex traits, adult CML risk may be modulated by other genetic markers besides MTHFR genes. Thus, fully elucidating the pathogenesis of CML would demand an investigation into the association and combined interaction of many gene variants with adult CML risk. Third, the included studies only focused on the Asian and Caucasian populations. Thus, further studies on a wider spectrum of subjects should be carried to investigate the role of these variants in different ethnicities. Last, this meta-analysis was based on unadjusted ORs and possible effect modifiers, such as demographics and other clinical characteristics, may influence the estimates of associations. The calculation of adjusted pooled ORs may provide more insight into the associations. Unfortunately, individual GAS did not provide uniform adjusted risk estimates. Thus, further well-designed GAS need to focus on exploring sources of heterogeneity. Despite these limitations, our study is the first comprehensive meta-analysis of all eligible studies on the associations between the MTHFR C677T and A1298C polymorphisms and adult CML risk.
In summary, the current meta-analysis indicates that the C allele of MTHFR A1298C may be associated with a decreased risk of adult CML, especially among Asians, while MTHFR C677T may not be associated with adult CML risk. Thus, MTHFR A1298C can probably be used with other genetic markers together to identify individuals at high risk for adult CML. However, due to the limitations of this study, these results should be interpreted with caution and still require future large-scale studies to confirm their accuracy. Moreover, considering that CML is a complex disease with a multifactorial etiology, the development of adult CML might be associated with gene-gene and gene-environment interactions, whose effects should be considered in future GAS and subsequent meta-analyses that may provide more conclusive evidence regarding the genetic susceptibility to adult CML.
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Gyurkocza B, Storb R, Storer BE, Chauncey TR, Lange T, et al. Nonmyeloablative allogeneic hematopoietic cell transplantation in patients with acute myeloid leukemia. J Clin Oncol. 2010;28:2859–67.
Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–302.
Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5:311–21.
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–56.
Quintas-Cardama A, Cortes JE. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81:973–88.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64.
Lucock M. Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Genet Metab. 2000;71:121–38.
Duthie SJ, Narayanan S, Blum S, Pirie L, Brand GM. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr Cancer. 2000;37:245–51.
Bailey LB, Gregory 3rd JF. Folate metabolism and requirements. J Nutr. 1999;129:779–82.
Wiemels JL, Smith RN, Taylor GM, Eden OB, Alexander FE, et al. Methylenetetrahydrofolate reductase (MTHFR) polymorphisms and risk of molecularly defined subtypes of childhood acute leukemia. Proc Natl Acad Sci U S A. 2001;98:4004–9.
Fodinger M, Horl WH, Sunder-Plassmann G. Molecular biology of 5,10-methylenetetrahydrofolate reductase. J Nephrol. 2000;13:20–33.
Duthie SJ. Folic acid deficiency and cancer: mechanisms of DNA instability. Br Med Bull. 1999;55:578–92.
Duthie SJ, Narayanan S, Brand GM, Pirie L, Grant G. Impact of folate deficiency on DNA stability. J Nutr. 2002;132:2444S–9S.
Krajinovic M, Lamothe S, Labuda D, Lemieux-Blanchard E, Theoret Y, et al. Role of MTHFR genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Blood. 2004;103:252–7.
Hanson NQ, Aras O, Yang F, Tsai MY. C677T and A1298C polymorphisms of the methylenetetrahydrofolate reductase gene: incidence and effect of combined genotypes on plasma fasting and post-methionine load homocysteine in vascular disease. Clin Chem. 2001;47:661–6.
Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;64:169–72.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, et al. Strengthening the reporting of genetic association studies (STREGA): an extension of the STROBE statement. Hum Genet. 2009;125:131–51.
Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25:603–5.
Jackson D, White IR, Riley RD. Quantifying the impact of between-study heterogeneity in multivariate meta-analyses. Stat Med. 2012;31:3805–20.
Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58.
van Houwelingen HC, Arends LR, Stijnen T. Advanced methods in meta-analysis: multivariate approach and meta-regression. Stat Med. 2002;21:589–624.
Sacks HS, Berrier J, Reitman D, Ancona-Berk VA, Chalmers TC. Meta-analyses of randomized controlled trials. N Engl J Med. 1987;316:450–5.
Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Comparison of two methods to detect publication bias in meta-analysis. JAMA. 2006;295:676–80.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315:629–34.
Barbosa CG, Souza CL, de Moura Neto JP, Arruda M, Barreto JH, et al. Methylenetetrahydrofolate reductase polymorphisms in myeloid leukemia patients from Northeastern Brazil. Genet Mol Biol. 2008;31:29–32.
Deligezer U, Akisik E, Dalay N. Genotyping of the MTHFR gene polymorphism, C677T in patients with leukemia by melting curve analysis. Mol Diagn. 2003;7:181–5.
Hur M, Park JY, Cho HC, Lee KM, Shin HY, et al. Methylenetetrahydrofolate reductase A1298C genotypes are associated with the risks of acute lymphoblastic leukaemia and chronic myelogenous leukaemia in the Korean population. Clin Lab Haematol. 2006;28:154–9.
Hussain SR, Naqvi H, Raza ST, Ahmed F, Babu SG, et al. Methylenetetrahydrofolate reductase C677T genetic polymorphisms and risk of leukaemia among the North Indian population. Cancer Epidemiol. 2012;36:e227–31.
Ismail SI, Ababneh NA, Awidi A. Methylenetetrahydrofolate reductase (MTHFR) genotype association with the risk of chronic myelogenous leukemia. Jordan Med J. 2009;43:8–14.
Jakovljevic K, Malisic E, Cavic M, Radulovic S, Jankovic R. Association between methylenetetrahydrofolate reductase polymorphism C677T and risk of chronic myeloid leukemia in Serbian population. Leuk Lymphoma. 2012;53:1327–30.
Jankovic RN, Jakovljevic K, Cavic M, Malisic E. Relation of methylenetetrahydrofolate reductase C677T polymorphism to chronic myeloid leukemia in Serbia. J Clin Oncol. 2011;29.
Kim HN, Kim YK, Lee IK, Yang DH, Lee JJ, et al. Association between polymorphisms of folate-metabolizing enzymes and hematological malignancies. Leuk Res. 2009;33:82–7.
Lordelo GS, Miranda-Vilela AL, Akimoto AK, Alves PC, Hiragi CO, et al. Association between methylene tetrahydrofolate reductase and glutathione s-transferase M1 gene polymorphisms and chronic myeloid leukemia in a Brazilian population. Genet Mol Res. 2012;11:1013–26.
Moon HW, Kim TY, Oh BR, Min HC, Cho HI, et al. MTHFR 677CC/1298CC genotypes are highly associated with chronic myelogenous leukemia: a case-control study in Korea. Leuk Res. 2007;31:1213–7.
Vahid P, Farnaz R, Zaker F, Farzaneh A, Parisa R. Methylenetetrahydrofolate reductase gene polymorphisms and risk of myeloid leukemia. Lab Med. 2010;41:490–4.
Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22:4632–42.
Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004;13:511–9.
Munafo MR, Flint J. Meta-analysis of genetic association studies. Trends Genet. 2004;20:439–44.
Bai JL, Zheng MH, Xia X, Ter-Minassian M, Chen YP, et al. MTHFR C677T polymorphism contributes to prostate cancer risk among Caucasians: a meta-analysis of 3511 cases and 2762 controls. Eur J Cancer. 2009;45:1443–9.
Collin SM, Metcalfe C, Zuccolo L, Lewis SJ, Chen L, et al. Association of folate-pathway gene polymorphisms with the risk of prostate cancer: a population-based nested case-control study, systematic review, and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2009;18:2528–39.
Boccia S, Boffetta P, Brennan P, Ricciardi G, Gianfagna F, et al. Meta-analyses of the methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and risk of head and neck and lung cancer. Cancer Lett. 2009;273:55–61.
Boccia S, Hung R, Ricciardi G, Gianfagna F, Ebert MP, et al. Meta- and pooled analyses of the methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer risk: a huge-GSEC review. Am J Epidemiol. 2008;167:505–16.
Dong X, Wu J, Liang P, Li J, Yuan L, et al. Methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer: a meta-analysis. Arch Med Res. 2010;41:125–33.
Eroglu A, Akar N. Methylenetetrahydrofolate reductase C677T polymorphism in breast cancer risk. Breast Cancer Res Treat. 2010;122:897–8.
Ding XP, Feng L, Ma L. MTHFR C677T polymorphism and ovarian cancer risk: a meta-analysis. Asian Pac J Cancer Prev. 2012;13:3937–42.
Chen H, Zhu J. C677T polymorphism of methylenetetrahydrofolate reductase may contribute to cervical cancer risk in complete over-dominant model. Med Hypotheses. 2013;80:679–83.
Guo LN. Methylenetetrahydrofolate reductase C677T polymorphism and cervical cancer risk: a meta-analysis. Asian Pac J Cancer Prev. 2012;13:2193–7.
Chen K, Jiang QT, He HQ. Relationship between metabolic enzyme polymorphism and colorectal cancer. World J Gastroenterol. 2005;11:331–5.
Robien K, Ulrich CM. 5,10-Methylenetetrahydrofolate reductase polymorphisms and leukemia risk: a huGE minireview. Am J Epidemiol. 2003;157:571–82.
Pereira TV, Rudnicki M, Pereira AC, Pombo-de-Oliveira MS, Franco RF. 5,10-Methylenetetrahydrofolate reductase polymorphisms and acute lymphoblastic leukemia risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1956–63.
Wang J, Zhan P, Chen B, Zhou R, Yang Y, et al. MTHFR C677T polymorphisms and childhood acute lymphoblastic leukemia: a meta-analysis. Leuk Res. 2010;34:1596–600.
Yan J, Yin M, Dreyer ZE, Scheurer ME, Kamdar K, et al. A meta-analysis of MTHFR C677T and A1298C polymorphisms and risk of acute lymphoblastic leukemia in children. Pediatr Blood Cancer. 2012;58:513–8.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Bin Li, Jian Zhang, and Lei Wang have the same contributions to this study and should be considered as co-first authors.
Rights and permissions
About this article
Cite this article
Li, B., Zhang, J., Wang, L. et al. MTHFR genetic polymorphisms may contribute to the risk of chronic myelogenous leukemia in adults: a meta-analysis of 12 genetic association studies. Tumor Biol. 35, 4233–4245 (2014). https://doi.org/10.1007/s13277-013-1554-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-013-1554-9