
Abstract
To determine the etiological factors of human colorectal cancer (CRC) we assessed the frequency and prognostic significance of hMLH1 and hMSH2 genes in conjunction with hMLH1 and hMSH2 protein expression in 30 Indian CRC patients. The protein expression and promoter methylation of hMLH1 and hMSH2; Mismatch Repair genes (MMR) were analyzed by immunohistochemistry and methylation-specific PCR (MSP), respectively. A loss of hMLH1 expression was recognized in 4(13.3 %) and loss of hMSH2 expression was recognized in 2(6.6 %) of 30 CRC cases whereas 50 % tumors showed reduced expression of hMLH1 and 33.3 % showed reduced expression of hMSH2 protein. One tumor showed a loss of both hMLH1 and hMSH2 expression. Normal nuclear staining pattern of hMLH1 and hMSH2 was observed in almost all the adjoining and normal mucosa. Promoter hypermethylation of the hMLH1 gene was detected in 15 of 30 CRC cases (50 %) and of hMSH2 gene was only in 3 of 30 CRC cases (10 %). No promoter methylation of hMLH1 and hMSH2 genes was observed in adjoining and normal mucosa. Combination of methylation of hMLH1 and hMSH2 gene was observed in two tumors (6.6 %). A significant correlation between histological grade of the tumor, methylation and expression of hMLH1 gene (p < 0.05) was observed. Normal expression of hMLH1 and hMSH2 was seen in all of the unmethylated tumors (100 %). Nuclear staining and promoter methylation of hMLH1 and hMSH2 did not significantly influence survival. hMLH1 methylation was common and was significantly correlated with loss of hMLH1 protein expression. In contrast, hMSH2 methylation was infrequent. These findings suggest that the inactivation of MMR gene expression probably via hypermethylation may lead to inactivation of their functions which finally leads to tumor aggressiveness and the immunostaining of hMLH1 protein can be used as a prognostic factor for determining the grade of the tumor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CRC is a major cause of cancer-associated morbidity and mortality worldwide. It is the second most prevalent cancer and affects men and women almost equally [1]. The epidemiologies of CRC have been studied in Indian population [2]. However, the molecular mechanisms involved in the pathogenesis have not been investigated in Indian CRC patients.
DNA methylation is the most studied epigenetic alteration. In higher-order eukaryotes, DNA is methylated only at cytosine located 5′ to guanosine in the CpG dinucleotide. This modification has important regulatory effects on gene expression, especially when involving CpG rich areas known as CpG islands, located in the promoter region of many genes. In cancer, DNA methylation of the promoter region of a normal tumor-suppressor gene leads to the aberrant silencing of its functions.
The development of human cancer is associated with genomic instability, which causes the accumulation of genetic changes that eventually result in the conversion of normal cells to malignant phenotypes. The human MMR system repairs DNA replication errors or physicochemical damage. Microsatellite regions are susceptible to mutation due to slippage of DNA polymerase during DNA replication. Failure to excise these errors may lead to frameshift mutations in many target genes [3]. MLH1, MSH2, MSH6, and PMS2 are major MMR genes implicated in genetic stability [4]. Genetic instability of microsatellite repeat sequences, i.e., microsatellite instability (MSI) is commonly seen in tumors associated with the hereditary nonpolyposis colorectal cancer syndrome (HNPCC) whereas it has also been observed in approximately 13 % of sporadic CRC [5-7]. In HNPCC patients, germ-line mutations have been identified in hMLH1 and hMSH2 genes [8]; however, somatic mutations in one of the DNA MMR genes have been reported in up to 26 % of sporadic MSI colorectal carcinoma [9-14].
Promoter region methylation has recently been demonstrated to be an important mechanism of gene inactivation in cancer [15-18]. MSI has been associated with hypermethylation of the hMLH1 promoter region in sporadic CRC cases [19-22]. However, in a significant subset of sporadic tumors with MSI no mutations of MMR genes could be identified [23-27] and it was speculated that non-mutational mechanisms or novel genes were responsible for the defect [26, 27]. Recent studies on sporadic colorectal carcinoma have found methylation of the promoter region of hMLH1 in 84–89 % of the tumors with MSI, whereas methylation of the promoter region of hMSH2 seems to be rare [28-31]. However, no data regarding the inactivation of these MMR genes in colorectal cancer are available from India.
Therefore, in the present study, to understand the etiology of human colorectal cancer in Indian patients, we performed an immunohistochemical and promoter methylation analysis for the two MMR genes, hMLH1 and hMSH2. We also examined the correlation between the promoter methylation and loss of expression. The relationship between methylation and immunohistochemical patterns of hMLH1/hMSH2 expression and the clinical and pathological features of colorectal adenocarcinoma as well as the correlation of these markers with clinical outcome was also analyzed in 30 CRC patients.
Material and methods
Study design
The study was a prospective analysis of 30 patients with histologically proven colorectal adenocarcinoma admitted to the Postgraduate Institute of Medical Education and Research, India between July 2004 and July 2006. Histological classification of the tumor types and stages was performed according to the World Health Organization classification method and the Tumor, Node, Metastasis System, respectively. Information on the clinicopathological features of CRC patients was obtained from hospital records. The study was reviewed and approved by the Institute Ethics Committee which allowed us to obtain tissue samples and all pertinent follow-up information. Only patients undergoing resectional surgery were included. Patients who had history of prior chemotherapy or radiotherapy, with inoperable tumors, family history of colorectal adenocarcinoma, or those with mucinous/signet cell carcinoma were excluded from the study. Follow-up end point was September, 2007. Apart from the description of the gross features of the tumor at the time of surgery, the rest of the colon was examined for any synchronous polyp or tumor. Fresh samples from tumor, adjoining (2–5 cm from the tumor) tissue and distant mucosa (5–10 cm from the main tumor mass) were taken from the resected colorectal specimen.
For histopathological analysis, freshly removed tissue samples were immediately fixed in 10 % buffered formalin for 24 h, embedded in paraffin and histopathological assessment was carried out to determine the tumor grade and invasion. Fresh tissues were snap frozen within 10–15 min of surgical removal and stored at −80 °C till further use. Each tissue for the molecular analysis was also assessed histologically by making a crushed smear to ensure the presence of tumors and only those samples, which contained >90 % of tumor cells were included for the final analysis. Similarly, the presence of adjoining and normal colorectal mucosa was also confirmed histologically before subjecting the tissue for further analysis. The normal mucosa was used as a control in each case.
Detection of promoter methylation of hMLH1 and hMSH2
For methylation analysis, all the specimens obtained during surgery procedure were treated with proteinase K and RNase. Each specimen was then subjected to DNA extraction using standard phenol–chloroform procedures [32]. The promoter methylation status of hMLH1 and hMSH2 gene was assessed by chemical treatment with sodium bisulfite and subsequent methylation-specific PCR (MSP) analysis according to the method of Herman et al. [33].
MSP for the hMLH1 and hMSH2 gene
Briefly, DNA (1 μg) in a volume of 50 μl was denatured by 0.2 M NaOH and incubated at 37 °C for 10 min; 30 μl of 10 mM hydroquinone (Sigma, St. Louis, USA) and 520 μl of 3 M Na bisulfite, pH 5.0 (Sigma, St. Louis, USA), both freshly prepared were added to denatured DNA solution. The tubes were then incubated at 50 °C/16 h. The bisulfite-treated DNA was purified using the wizard DNA clean up system (Promega, Madison, WI, USA). Modification was completed by 0.3 M NaOH treatment for 5 min at room temperature, followed by ethanol precipitation, DNA was resuspended in distilled water and stored at −20 °C. The bisulfite modified DNA was PCR amplified by using primers specific for methylated CpG and unmethylated regions of hMLH1 and hMSH2 promoters. The PCR mixture contained 1× PCR buffer (16.6 mM ammonium sulfate/67 mM Tris, pH 8.8/6.7 mM MgCl2/10 mM 2-mercaptoethanol), dNTPs (each at 2 mM), 10 pmol of each primer and bisulfite-treated DNA (~50 ng) or unmodified DNA (50–100 ng) in a final volume of 25 μl. Primer sequences of hMLH1 for unmethylated reaction were 5′ TTT TGA TGT AGA TGT TTT ATT AGG GTT GT 3′ (sense) and 5′ ACC ACC TCA TCA TAA CTA CCC ACA 3′ (antisense) and for methylated reaction were 5′ ACG TAG ACG TTT TAT TAG GGT CGC 3′ (sense) and 5′ CCT CAT CGT AAC TAC CCG CG 3′ (antisense). Primer sequences of hMSH2 for unmethylated reaction were 5′ GGT TGT TGT GGT TGG ATG TTG TTT 3′ (sense) and 5′ CAA CTA CAA CAT CTC CTT CAA CTA CAC CA 3′ (antisense) and for methylated reaction were 5′ TCG TGG TCG GAC GTC GTT C 3′ (sense) and 5′ CAA CGT CTC CTT CGA CTA CAC CG 3′ (antisense). PCR specific for unmodified DNA also included 5 % DMSO. Reactions were hot started at 95 °C for 5 min before the addition of 1.5 units of Taq DNA polymerase (Roche, GmbH, Germany). PCR amplification of the modified DNA samples consisted of 1 cycle of 95 °C for 5 min; 40 cycles of 95 °C for 30 s, 59 °C for 1 min, and 72 °C for 1 min; and 1 cycle of 72 °C for 5 min. PCR products amplified by unmethylated and methylated primers were 124 and 115 bp respectively for hMLH1 and 143 and 132 bp, respectively, for hMSH2. The PCR products were analyzed by agarose gel electrophoresis. DNA treated in vitro with CpG methylase MSssI (Sss1 methyltransferase, New England Biolabs, Ipswich, MA, USA) was used as a positive control for methylated alleles of these genes. Controls without DNA were performed for each set of PCR. Each MSP was repeated at least three times. Analysis of DNA methylation was performed in the absence of knowledge of the hMLH1 and hMSH2 expression status in all of the experiments.
Analysis of protein expression: immunohistochemistry assay
The immunohistochemistry (IHC) was performed on histology sections taken from the surgically resected specimens. All the tissues fixed in formalin were processed to make paraffin blocks. Five-micrometer sections of paraffin embedded tissues were mounted on slides coated with poly-l-lysine (Sigma, St. Louis, USA). The sections were deparaffinized by heating at 60 ºC, followed by serial passages though few changes of xylenes and graded alcohol (100, 95, and 70 %). The endogenous peroxidase activity was blocked by incubating the sections with the blocking solution (0.03 % H2O2/methanol) for 20 min. The antigenic sites were unmasked by means of conventional household psi pressure cooker treatment for 15 min in 10 mM citrate buffer (pH 6.0). Antibodies used were MLH1 (N-20) sc-581 (1:20) to detect hMLH1 protein and MSH2 (N-20) sc-494 (1:40; Santa Cruz Biotechnology Inc. CA, USA) to detect hMSH2 protein using a peroxidase-labeled streptavidin–biotin technique. Sections were incubated for 3 h at room temperature. Negative controls were sections treated by the same techniques but without the primary antibody. Two slides per patient sample were immunostained in separate runs. Slides were scored by a consultant pathologist using light microscopy.
The normal staining pattern for hMLH1 and hMSH2 is nuclear. Tumor cells that exhibited an absence of nuclear staining in the presence of non-neoplastic cells and infiltrating lymphocytes with nuclear staining were considered to have an abnormal pattern. Staining results were examined without knowledge of the status of the molecular analyses.
Scoring of immunostaining
The immunohistochemistry results were scored by taking percentage positivity and intensity of staining into account. A score of 0 to 3 for stain intensity was assigned: an intensity score of 0 = no staining, 1 = weak positivity, 2 = moderate positivity and 3 = strong positivity was given. The total IHC score was calculated as: IHC score = %age of positivity × intensity score.
Statistical methods
Pearson's χ 2 test was performed to analyze the relationship between hMLH1 and hMSH2 methylation status with their respective protein expression and with each of the clinicopathological parameters. Mann–Whitney U test was performed to determine the relationship between the expression of hMLH1 and hMSH2 and each of the clinicopathological parameters. Comparisons of the different groups were performed using the ANOVA followed by post-hoc test. The overall survival (OS) and disease-free survival (DFS) were estimated by the Kaplan–Meier method and the logrank test was used to evaluate the difference between survival of the patients with and without methylation and expression of hMLH1 and hMSH2.
Results
Clinical profile
There were 30 patients (20 males) with age range from 24–90 years (median age 56 years). Eighteen (60 %) patients had the tumor in distal colon, and 12 (40 %) in the proximal colon. None of the patients had a synchronous adenoma or carcinoma. The median length of the tumors was 5 cm (range 2–10 cm). There were four patients (13.3 %) in stage I, 18 (60 %) in stage II, 6 (20 %) in stage III and 2 (6.6 %) in stage IV disease. Metastasis was found in eight patients with distant metastasis in liver (n = 2) and lymph node (n = 6). None of the patients had a family history of CRC or any other kind of malignancy. All tumors were adenocarcinomas and on histology, 6 were well differentiated, 21 moderately differentiated, and 3 poorly differentiated adenocarcinomas.
Promoter methylation analysis of hMLH1 and hMSH2
Methylation status of hMLH1 and hMSH2 genes was evaluated in 30 colorectal tumors, adjoining and normal mucosa. Out of 30 cases of CRC, promoter methylation of hMLH1 and hMSH2 genes was observed in 15 (50 %) and 3 (10 %) tumors, respectively. As the tumor samples also contain normal cells, so the amplification of the unmethylated sequence was observed in all tumors. In colorectal adjoining and normal mucosa only unmethylated band of hMLH1 and hMSH2 genes were present. No promoter methylation of hMLH1 and hMSH2 genes was observed in adjoining and normal mucosa (Fig. 1). Combination of methylation of hMLH1 with hMSH2 genes was observed in 2 (6.6 %) tumors.

Depicts promoter methylation analysis of hMLH1 and hMSH2 in representative cases of colorectal cancer (CRC) in normal (N), adjoining (A) and tumor (T). hMLH1 (U) shows unmethylated PCR products for hMLH1 gene and hMLH1 (M) shows methylated PCR products for hMLH1gene. Similarly, hMSH2 (U) shows Unmethylated PCR products for hMSH2 gene and hMSH2 (M) shows methylated PCR products for hMSH2 gene
Protein expression of hMLH1
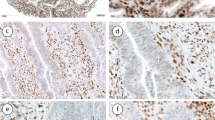
In colorectal adenocarcinoma 26 (86.6 %) tumors showed hMLH1 protein expression. Complete loss of hMLH1 protein expression was observed in 4 (13.3 %) tumors. Out of 26 positively stained tumors hMLH1 expression was observed weak in 15, moderate in 10 and strong in 1 tumor (Fig. 2a–b). hMLH1 protein showed immunoreactivity in all the 30 adjoining and normal mucosa (Fig. 2c–d). The expression of hMLH1 protein was weak in 6, moderate in 20 and strong in 4 cases of adjoining mucosa. There was a significant difference in the expression of hMLH1 in adjoining and normal mucosa as compared to tumor at nuclear level (p = 0.001).

Depicts protein expression of hMLH1in representative case of colorectal cancer (CRC). a and b show protein expression of hMLH1 in tumor tissues; c and d show protein expression of hMLH1 in adjoining and normal mucosa
Protein expression of hMSH2
hMSH2 protein showed immunoreactivity in 28 tumors, while loss of expression was observed in 2 tumors (6.6 %). Out of 28 positively stained tumors, weak expression was observed in 10 tumors, moderate in 17, and strong in 1 tumor (Fig. 3a–b). In adjoining and normal mucosa, positive immunostaining was present in all the 30 samples (Fig. 3c–d). Weak expression of hMSH2 protein was observed in 8, moderate in 17, and strong in 5 samples. There was a significant difference in the expression of hMSH2 protein between tumor and normal mucosa at nuclear level (p = 0.029).

Protein expression of hMLH1in representative case of colorectal cancer (CRC). a and b show protein expression of hMSH2 in tumor tissues; c and d show protein expression of hMSH2 in adjoining and normal mucosa
Clinicopathological correlation with promoter methylation and expression
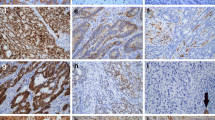
Methylation and expression of the hMLH1 and hMSH2 genes had no correlation with clinicopathological factors (p > 0.05) including, TNM stage, Duke's stage, smoking/alcohol consumption, metastasis, tumor site, pre- and postoperative serum CEA levels, age, etc. A significant correlation was observed between the histological grade of the tumor and expression of hMLH1 gene (p < 0.05) (Fig. 4).

Depicts correlation of hMLH1 protein expression and histological grade of tumor in representative case of colorectal cancer (CRC)
Correlation of promoter methylation with the protein expression
The protein expression of hMLH1 was compared to promoter methylation of hMLH1 and the results are summarized in Table 1. Out of the 26 colorectal adenocarcinoma positive for hMLH1 protein expression, 11 had methylation in the promoter region of hMLH1 gene. Out of 11 methylated samples, 7 had reduced expression and 4 had moderate expression. However, four tumors totally negative for hMLH1 expression also had methylation in the promoter region. There was a significant effect of hMLH1 gene methylation on the expression of hMLH1 protein (p = 0.002).
Similarly, the protein expression of hMSH2 was compared with respect to their respective gene methylation and the results are summarized in Table 2. In case of 28 colorectal adenocarcinomas positive for hMSH2 protein expression, 1 had methylation in the promoter region of hMSH2 gene. However, two tumors totally negative for hMSH2 expression also had methylation in their promoter regions. However, one case showed a loss of both hMLH1 and hMSH2 expression.
Survival analysis
A follow-up data of the patients enrolled for this study was obtained for 29 patients. At the last follow-up (September, 2007), four patients (13.3 %) died, 4 patients (13.3 %) showed the recurrence of disease with distant metastasis in 2 patients (6.6 %) and 21 patients (70 %) were alive without disease.
The association of methylation and expression of hMLH1 and hMSH2 with disease-free and overall survival was analyzed. Disease-free survival was defined as the time from the date of surgical resection of the tumor to the date of recurrence of the disease. The median follow-up period was 27 months with a range of 8–39 months (mean = 25.5 ± 8.14). Overall survival was defined as the time from the date of diagnosis of CRC to the date of last follow-up. The median follow-up period was 27.5 months with a range of 8–39 months (mean = 26.36 ± 8.19). By Kaplan–Meier logrank survival analysis it was found that the methylation and expression of hMLH1 and hMSH2 has no effect on the survival of the patients with colorectal adenocarcinomas (p > 0.05).
Discussion
There are two major mechanisms of gene inactivation. First is the genetic mechanism, e.g., the aberration of DNA structure such as homozygous deletion or intragenic mutation resulting in the gene inactivation and second is the epigenetic mechanism, e.g., the methylation at position 5′ of cytosine residue leading to the lack of gene expression, while the structure and the product of the gene remain unchanged.
The human DNA repair system plays an important role in reducing mutations and maintaining genomic stability. The MMR genes hMLH1 and hMSH2 are integral components of the DNA mismatch repair pathway. Defective DNA mismatch repair is most commonly associated with the functional loss of hMLH1 and hMSH2 genes and results in the mutator phenotype characterized by MSI in CRC. Aberrant methylation, which can result in the transcriptional silencing of the target gene, was frequently found in the hMLH1 and hMSH2 alterations in colorectal cancer. In the present study, MSP assays were used to examine the promoter methylation of hMLH1 and hMSH2 genes in tumor, adjoining and normal mucosa. The methylation frequencies of hMLH1 were observed in 50 % and of hMSH2 in 10 % of tumors, whereas no promoter methylation of hMLH1 and hMSH2 genes was observed in adjoining and normal mucosa. The hypermethylation of hMSH2 gene was quite low in this population study, consistent with the previous study in Asian population [29]. However, majority of the studies have reported absence of promoter methylation of hMSH2 gene [28, 30]. In a study, Herman et al. [30] reported hypermethylation of hMLH1 in 84 % of MSI+ sporadic CRCs and suggested that methylation of the hMLH1 promoter is an epigenetic event that plays a causal role in the MMR defect in many MSI+ cancers. In another study, Miyakura et al. reported methylation of hMLH1 gene promoter in 88.9 % of CRCs in Japanese [31]. The difference in frequency of methylation may be due in part to differences in the type or stage of tumors analyzed in the contrasting studies. It is also possible that geographical/environmental factors also account for the frequent alterations of hMLH1 and hMSH2. Majority of the studies reported methylation of hMLH1 and hMSH2 gene and correlated with MSI status but in the present study MSI was not analyzed.
Further, the correlation of hMLH1 and hMSH2 gene methylation with different clinicopathological factors was analyzed and it was observed that hypermethylation of hMLH1 and hMSH2 gene did not correlate with age, metastases, pre- and postoperative serum CEA level, stage, and tumor size. A significant positive correlation between the methylation of hMLH1 and the histological grade of the tumor (p = 0.040) was observed. Frequent methylation of hMLH1 gene was found to be present in poorly differentiated colorectal carcinoma suggesting that inactivation of the gene due to methylation leads to tumor aggressiveness.
The immunohistochemical analysis of hMLH1 and hMSH2 protein was also done to evaluate the prognostic significance of MMR status in tumor tissues, adjoining and normal mucosa of sporadic CRC patients. Almost all the tumors expressed both hMLH1 and hMSH2 proteins. Complete loss of hMLH1 expression was observed in four tumors (13.3 %) and of hMSH2 in two tumors (6.6 %) whereas 50 % tumors showed reduced expression of hMLH1 and 33.3 % showed reduced expression of hMSH2 protein. These results are consistent with other studies [34]. The normal nuclear staining pattern of hMLH1 and hMSH2 was observed in majority of adjoining and normal mucosa whereas some cases of adjoining mucosa showed reduced expression of hMLH1 and hMSH2 protein. Hammed et al. [35] have reported that 60 % of the colorectal tumors demonstrated normal expression of both hMLH1 and hMSH2 protein and 27 % tumors did not express hMLH1 and 13 % hMSH2 proteins. In another study, Lanza et al. [34] reported that MSS or MSI-L tumors showed normal hMLH1/hMSH2 expression by immunohistochemistry whereas MSI-H carcinomas showed complete loss of hMLH1/hMSH2 protein expression. However, in the present study, a highly significant correlation was found between hMLH1 and hMSH2 gene methylation and loss/reduced protein expression (p < 0.01) in tumors from colorectal cancer patients, suggesting that promoter methylation is the predominant mechanism by which these two genes are silenced and might affect the protein expression in sporadic CRC. In addition, some tumors also expressed hMLH1 even with the methylation in their promoter regions. This may be due to the reason that in such cases only one allele is methylated and the remaining unmethylated allele would be expected to produce a sufficient amount of hMLH1 protein, resulting in proficient mismatch repair. Alternatively, it might be possible that methylation of other CpG sites is important in silencing the hMLH1 gene or otherwise full methylation in a wide region of hMLH1 promoter is necessary for gene silencing of hMLH1. However, few cases also showed reduced expression of hMSH2 protein without the promoter methylation in their promoter region which indicates that methylation of the specific sites examined may be important but may not be sufficient for gene inactivation and a broad analysis of all the CpG sites are required.
Further, the correlation of hMLH1 and hMSH2 protein expression with different clinicopathological factors was analyzed and it was observed that loss/reduced expression of hMLH1 and hMSH2 protein did not correlate with any of the clinicopathological factors examined except the degree of differentiation of the tumor. There was significant association between hMLH1 protein expression and tumor grade. The expression of hMLH1 protein was reduced in poorly differentiated adenocarcinomas as compared to the well-differentiated adenocarcinomas. A similar observation was reported with the methylation of hMLH1 gene and tumor grade which suggests that loss/reduction in the expression of hMLH1 as a result of hypermethylation increases as the grade of the tumor increases. So, the frequent occurrence of promoter methylation of the hMLH1 gene and loss/reduced expression of hMLH1 protein in poorly differentiated carcinomas indicates its potential use as a diagnostic marker in CRC. This should be confirmed in larger patient subsets.
Finally, the clinical implications of various molecular changes in the patient’s cohort was studied to establish, whether the observed changes affect the disease free survival and the overall survival. In the present study, no correlation could be established for the methylation and expression of hMLH1 and hMSH2 genes to the survival using univariate analysis.
All reported studies have conserved the adenoma → carcinoma sequence and reported the presence of genetic alterations in adenomas, a precursor lesion that finally develops to carcinoma. But only a very small number of the Asian patient population have adenomas; therefore, the present study included adjoining mucosa so as to determine the initial changes in CRC. But in case of adjoining mucosa, no major changes were observed which suggested that there are no changes near the tumor region and the process of tumorigenesis is restricted to a limited area.
An important aspect of this study is that for the first time, two major MMR genes have been analyzed at both genetic and expression level in CRC in relation to pathophysiology and prognosis in small Indian patient cohort. The limitation of the study is the small size of the cohort. As this was the pilot study on Indian population, we need more patients from different parts of the country to validate our findings.
In conclusion, our study demonstrated that majority of CRC tissues expressed MMR proteins hMLH1 and hMSH2 and the low or reduced expression of these proteins among CRC tissues probably via hypermethylation may leads to inactivation of their functions which finally leads to tumor aggressiveness and the immunostaining of hMLH1 protein can be used as a prognostic factor for determining the grade of the tumor. Although the cases in the present study were not enough to draw definite conclusions, however, it may be suggested that the promoter hypermethylation of hMLH1 and hMSH2 genes may be one of the causes of inactivation of these genes in sporadic CRC.
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108.
Sung JJ, Lau JY, Goh KL, Leung WK, Asia Pacific Working Group on Colorectal Cancer. Increasing incidence of colorectal cancer in Asia: implications for screening. Lancet Oncol. 2005;6:871–6. doi:10.1016/S1470-2045(05)70422-8.
Bacani J, Zwingerman R, Di Nicola N, Spencer S, Wegrynowski T, Mitchell K, et al. Tumor microsatellite instability in early onset gastric cancer. J Mol Diagn. 2005;7(4):465–77.
Gologan A, Sepulveda AR. Microsatellite instability and DNA mismatch repair deficiency testing in hereditary and sporadic gastrointestinal cancers. Clin Lab Med. 2005;25(1):179–96.
Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338:1481–7.
Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, et al. Science. 1993;260:812–6.
Thibodeau SN, Bren G, Schaid D. Science. 1993;260:816–9.
Peltoma¨ki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–58.
Borresen AL, Lothe RA, Meling GI, Lystad S, Morrison P, Lipford J, et al. Somatic mutations in the hMSH2 gene in microsatellite unstable colorectal carcinomas. Hum Mol Genet. 1995;4:2065–72.
Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet. 1995;9:48–55.
Moslein G, Tester DJ, Lindor NM, Honchel R, Cunningham JM, French A, et al. Microsatellite instability and mutation analysis of hMSH2 and hMLH1 in patients with sporadic, familial and hereditary colorectal cancer. Hum Mol Genet. 1996;5:1245–52.
Bubb VJ, Curtis LJ, Cunningham C, Dunlop MG, Carothers AD, Morris R, et al. Microsatellite instability and the role of hMSH2 in sporadic colorectal cancer. Oncogene. 1996;12:2641–9.
Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996;56:4836–40.
Wu Y, Nystrom Lahti M, Osinga J, Looman MW, Peltomaki P, Aaltonen L, et al. MSH2 and MLH1 mutations in sporadic replication error-positive colorectal carcinoma as assessed by two-dimensional DNA electrophoresis. Gene Chromosome Canc. 1997;18:269–78.
Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7.
Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 59 CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–92.
Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–96.
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91:9700–4.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–5.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11.
Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–60.
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95:8698–702.
Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, et al. Cancer Res. 1996;56:4836–40.
Shin KH, Shin JH, Kim JH. Park JG Mutational analysis of promoters of mismatch repair genes hMSH2 and hMLH1 in hereditary nonpolyposis colorectal cancer and early onset colorectal cancer patients: identification of three novel germ-line mutations in promoter of the hMSH2 gene. Cancer Res. 2002;62(1):38–42.
Yamada K, Zhong X, Kanazawa S, Koike J, Tsujita K, Hemmi H. Oncogenic pathway of sporadic colorectal cancer with novel germline missense mutations in the hMSH2 gene. Oncol Rep. 2003;10(4):859–66.
Jeong SY, Shin KH, Shin JH, Ku JL, Shin YK, Park SY, et al. Microsatellite instability and mutations in DNA mismatch repair genes in sporadic colorectal cancers. Dis Colon Rectum. 2003;46(8):1069–77.
Wu Y, Nystrom-Lahti M, Osinga J, Looman MW, Peltomaki P, Aaltonen LA, et al. Gene Chromosome Canc. 1997;18:269–78.
Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58(15):3455–60.
Kim HC, Kim CN, Yu CS, Roh SA, Kim JC. Methylation of the hMLH1 and hMSH2 promoter in early-onset sporadic colorectal carcinomas with microsatellite instability. Int J Colorectal Dis. 2003;18(3):196–202.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–5.
Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, et al. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterol. 2001;121(6):1300–9.
Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. p. 6.4–6.12.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi:10.1073/pnas.93.18.9821].
Lanza G, Gafa R, Santini A, Maestri I, Guerzoni L, Cavazzini L. Immunohistochemical test for MLH1 and MSH2 expression predicts clinical outcome in stage II and III colorectal cancer patients. J Clin Oncol. 2006;24(5):2359–67.
Hameed F, Goldberg PA, Hall P, Algar U, van Wijk R, Ramesar R. Immunohistochemistry detects mismatch repair gene defects in colorectal cancer. Colorectal Dis. 2006;8(5):411–7.
Acknowledgments
We acknowledge Dr J. D. Wig and the staff of OT and the Department of Exp. Med. and Biotech for helping in the collection of samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Malhotra, P., Anwar, M., Kochhar, R. et al. Promoter methylation and immunohistochemical expression of hMLH1 and hMSH2 in sporadic colorectal cancer: a study from India. Tumor Biol. 35, 3679–3687 (2014). https://doi.org/10.1007/s13277-013-1487-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-013-1487-3