
Abstract
Transforming growth factor beta (TGF-β) induces epithelial–mesenchymal transition (EMT) accompanied by cellular differentiation and migration. Despite extensive transcriptomic profiling, identification of TGF-β-inducible, EMT-specific genes during metastatic progression of lung cancer remains elusive. Here, we functionally validate a previously described post-transcriptional pathway by which TGF-β modulates expression of interleukin-like EMT inducer (ILEI), and EMT itself. We show that poly r(C)-binding protein 1 (PCBP1) binds ILEI transcript and repress its translation. TGF-β activation leads to phosphorylation at serine-43 of PCBP1 by protein kinase Bβ/Akt2, inducing its release from the ILEI transcript and translational activation. Modulation of hnRNP E1 expression modification altered TGF-β-mediated reversal of translational silencing of ILEI transcripts and EMT. Furthermore, ILEI could induce, as well as maintain, CD24lowCD44high subpopulation in A549 cells treated with TGF-β, which might explain its capability to induce metastatic progression. These results thus validate the existence of an evolutionary conserved TGF-β-inducible post-transcriptional regulon that controls EMT and subsequent metastatic progression of lung cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite rapid advances in drug development and surgical procedures, lung cancer is the leading cause of cancer death worldwide in both men and women [1], with an estimated 1.4 million deaths each year [2]. The alarming nature of these numbers is further highlighted by the fact that lung cancer death is greater than the next three most common cancers combined inclusive of colon, breast, and prostate cancer. Surgery is still considered the best option for non-small cell lung cancer (NSCLC) treatment. However, lung cancer is not usually diagnosed until advanced stage, and fewer than 25 % of NSCLC patients are considered as candidates for surgical therapy [3]. Chemotherapy is also not a viable option because whereas a large proportion of lung cancer patients are intrinsically resistant to chemotherapy referred as intrinsic resistance, other patients eventually develop acquired resistance even after combination therapy, although they are initially sensitive to chemotherapy [4]. Indeed, metastasis and therapeutic resistance are the major causes of failure in cancer treatment.
Epithelial–mesenchymal transition (EMT) is a core mechanism underlying tumor metastasis [4, 5], and it is postulated that epithelial cancer cells revert to an embryonic state during the invasive phase of this process, recapitulating the developmental switch from a polarized, epithelial phenotype to a highly motile mesenchymal phenotype [5]. The molecular mechanisms that induce and drive the cellular conversions of EMT have long been the focus of intense investigation. The vast majority of these studies have primarily focused on transcriptional regulation associated with EMT, revealing important roles for the Snail, Twist, and Zeb family of transcription factors and the signaling pathways that activate them [5, 6]. However, recent findings have revealed that vast, coordinated changes in post-transcriptional regulatory networks occur during EMT and that they profoundly alter cellular phenotypes and behaviors [7–9]. The impact of post-transcriptional control in the process of EMT, both by microRNAs (miRNAs) and RNA-binding proteins, is slowly evolving [7, 8, 10–15]; however, not much is known about post-transcriptional regulation of EMT and metastasis during lung cancer progression. Post-transcriptional regulation of gene expression during lung cancer pathogenesis and progression has been elucidated at the level of different miRNAs [16], but nothing is known about the RNA-binding proteins or the signaling mechanisms that dictate such regulation.
Previous work [7, 10, 12, 17] has shown that regulation of gene expression at the post-transcriptional level plays an indispensable role in transforming growth factor beta (TGFβ)-induced EMT. A transcript-selective translational regulatory pathway in which a ribonucleoprotein [messenger ribonucleoprotein particle (mRNP)] complex, consisting of poly r(C)-binding protein 1 (PCBP 1), binds to a 3′-UTR regulatory BAT (TGF-β activated translation) element and silences translation of Dab2 and ILEI messenger RNAs (mRNAs), which are involved in mediating EMT [7]. ILEI has been indicated initially in autosomal recessive nonsyndromic hearing loss locus 17 (DFNB17) and belongs to the FAM3A-D gene family [7]. It was shown that TGFβ activates a kinase cascade terminating in the phosphorylation of hnRNP E1 by isoform-specific stimulation of protein kinase Bβ/Akt2, inducing the release of the mRNP complex from the 3′-UTR element, in turn, resulting in the reversal of translational silencing and increased expression of transcripts that mediates EMT [7, 10, 17]. However, most of these experiments were conducted in normal mouse mammary epithelial cells, and no evidence exist that this pathway is actually functional during metastatic progression of human solid cancers. Hence, the objective of the current study was to functionally validate this pathway in an in vitro model using human lung cancer cell line.
Methods
Reagents
Primers and oligos were purchased from Integrated DNA Technologies (USA). Mouse α-hnRNP E1 was from Novus Biologicals (Oakville, ON, Canada), rabbit α-hnRNP E1 was from Lifespan Biosciences (Seattle, WA, USA). Antibodies against E-cadherin, vimentin, and Hsp90 were purchased from Cell Signaling Technology (Danvers, MA, USA). α-FAM3C antibody was obtained from Abcam (Cambridge, MA, USA), and α-phospho-serine (clone PSR-45) antibody from Sigma-Aldrich (India). Normal mouse and rabbit IgG were purchased from Santa Cruz Biotechnology (Shanghai, China).
Cell culture and treatments
A549 cells were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (FBS; Lonza, Heidelberg, Germany) and antibiotics/antimycotics (Life Technologies). A549 cells were cultured in Dulbecco's modified Eagle's medium medium (DMEM), containing 10 % FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Cells were kept at 37 °C, under a humidified atmosphere of 5 % CO2 in air. Where indicated, A549 cells were treated with TGF-β1 (R&D Systems, Minneapolis, MN, USA) at a final concentration of 5 ng/ml for the indicated time frame. Where indicated, ±TGF-β-treated cells were treated with MG-132 (10 μg/ml; Sigma-Aldrich) for up to 4 h.
Plasmids construction, transfection and stable clone generation
The human interleukin-like EMT inducer (ILEI) and overexpressing construct and poly r(C)-binding protein 1 (PCBP1) short hairpin RNA (shRNA), a pool of four target-specific 29mer, was brought from Thermo Fisher Scientific (Waltham, MA, USA). The human ILEI siRNA, a pool of three target-specific 19–25 nt. siRNAs, and control siRNA-A, a non-targeting 20–25 nt. siRNA, were brought from Santa Cruz Biotechnology. A549 cells (4 × 104) were transfected with Lipofectamine LTX (Life Technologies) with separate plasmids as indicated. Forty-eight hours after transfection, cells were split 1:10 and subsequently selected with G418 (1,000 μg/ml; Life Technologies) until distinct colonies were observed (approximately 2 weeks). The clones were pooled and used for subsequent analyses. Indicated PCBP1 shRNA and ILIE siRNA were transfected at 30 nM concentration.
Isolation of RNA and Northern blot
Isolation of total RNA and northern blot and RT-PCR was done as described previously [18]. The ILEI probe for the Northern blot was generated by PCR using forward primer 5′-ACACAGCTGCACGTTCTACA-3′ and reverse primer 5′-TCCAGGCAGATTTTGGGTCC-3′. The control cyclophilin (1B15) probe was generated through in vitro transcription of template obtained from Life Technologies.
Preparation of whole cell lysates, immunoblot analysis, and immunoprecipitation
For immunoblot analysis, cells were lysed in buffer containing 25 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 % NP-40, and 5 % glycerol containing complete, Mini protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). Whole cell lysates (10 μg) were resolved on a NuPAGE 4-20 % gel (Life Technologies), transferred to an Immobilon PVDF membrane (Millipore, Billerica, MA, USA), and probed with indicated antibodies. The blots were imaged using ECL Plus Western blotting substrate (Pierce, Rockford, IL, USA) and HyBlot CL autoradiography film (Denville Scientific, Metuchen, NJ, USA). Immunoprecipitation was performed as described previously [19].
Phenotypic analyses
Cells were washed once with phosphate-buffered saline (PBS) and then harvested with 0.05 % trypsin/0.025 % EDTA. Detached cells were washed with PBS containing 1 % FCS and 1 % bovine serum albumin (BSA;wash buffer) and resuspended in the wash buffer (106 cells/100 μl). Combinations of fluorochrome-conjugated monoclonal antibodies obtained from BD Biosciences (San Diego, CA, USA) against human CD44 (FITC cat. #555478) and CD24 (PE cat. #555428) or their respective isotype controls were added to the cell suspension at concentrations recommended by the manufacturer and incubated at 4 °C in the dark for 30 to 40 min. The labeled cells were washed in the wash buffer, then expressions of CD24 and CD44 were determined by flow cytometry using a FACSCalibur system (BD Biosciences), equipped with the filter set for quadruple fluorescence signals. All experiments were repeated at least three times and one representative result is represented. Data was analyzed using FlowJo version 9 (Tree Star, Ashland, OR). Analyses logic and coincidence decisions for identifying the CD24lowCD44high events were chosen for highest purity.
In vitro mammosphere formation assay
In vitro mammosphere formation was assessed in parental A549 cells or A549 cell line variants in which ILEI was being stably overexpressed or stably silenced. The aforementioned A549 cell types were plated at 20,000 cells/ml in serum-free, growth factor enriched conditions in 6-well ultra-low attachment plates in serum-free MEM supplemented with 20 ng/ml bFGF, 20 ng/ml EGF and B27 (Life Technologies). Suspension cultures were incubated for 10 days before colonies were counted with an automated colony counter (Oxford Optronix, Oxford, UK).
Results
TGF-β induces EMT of A549 cells
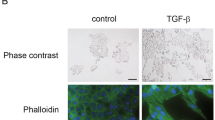
We initially wanted to determine if TGF-β treatment induces EMT in A549 cells. A549 cells showed distinct morphological changes associated with EMT 24 h after treated with TGF-β (data not shown). This was accompanied with a concomitant downregulation of the epithelial cell marker, E-cadherin and upregulation of the mesenchymal cell marker, vimentin (Fig. 1). This further suggested that TGF-β treatment could effectively induce EMT of A549 cells.

TGF-β induces EMT in A549 cells. Immunoblot (blot) analysis examining E-cadherin and vimentin protein levels in A549 cells treated with TGF-β for 24 h. The blot was stripped and reprobed with Hsp90 to confirm equal loading
ILEI protein expression is induced post-transcriptionally in A549 cells after TGF-β treatment
We next wanted to investigate if TGF-β-mediated induction of EMT in A549 cells is accompanied by a post-transcriptional induction of ILEI protein synthesis, as previously elucidated in murine mammary epithelial cells [7]. Northern blot was performed to explore the temporal relationship between steady state expression levels of ILEI mRNA and protein in TGF-β-treated A549 cells. A significant increase was observed in expression of ILEI protein (Fig. 2B) without a concomitant increase in its mRNA (Fig. 2A). However, unstimulated cells, despite having abundant ILEI mRNA, had low levels of ILEI protein (Fig. 2B). Since the steady-state levels of ILEI transcript was similar in A549 cells, irrespective of TGF-β treatment, we next determined if the difference in ILEI protein expression levels was attributable to differential proteasomal degradation in the two conditions. As shown in Fig. 2C, treatment with the proteasomal inhibitor MG-132, did not result in an induction of ILEI protein levels in TGFβ-untreated A549 cells.

TGF-β translationally upregulates ILEI expression. A Northern blot analysis examining ILEI expression levels in A549 cells treated with TGF-β for 24 h. The blot was stripped and reprobed with cyclophilin (1B15) to confirm equal loading. B Immunoblot (blot) analysis examining ILEI protein levels in A549 cells treated with TGF-β for 24 h. The blot was stripped and reprobed with Hsp90 to confirm equal loading. C MG-132 treatment for indicated time points of A549 cells ± TGF-β (24 h) analyzing the de novo rate of ILEI degradation
TGF-β induces serine-43 phosphorylation of PCBP1
Cell lysate from TGF-β treated A549 cells was harvested and phosphorylation of PCBP1 and serine was determined by immunoprecipitation. As shown in Fig. 3A, in the absence of TGF-β treatment, only baseline phosphorylation of serine-43 was observed. However, significant increase in PCBP1 phosphorylation was observed after TGF-β treatment, with peak of phosphorylation at 6 h after TGF-β treatment (Fig. 3A). We next made two variants of the parental A549 cell line:– one overexpressing PCBP1 (A549/PCBP1) and one where PCBP1 was knocked down (A549/PCBP1 −/−); Fig. 3B). The two variant cell lines along with the parental cell line were treated with TGF-β for up to 24 h, at which time the cells were harvested to prepare whole cell lysates. Overexpression of PCBP1 in A549 cells completely inhibited ILEI translation, as evident from lack of ILEI protein even after 24 h of stimulation with TGF-β (Fig. 3C). The role of PCBP1 in ILEI expression was further proved by the fact that in the A549/PCBP1−/− cells there was constitutive expression of ILEI, even without TGFβ stimulation.

A IB analysis of immunoprecipitates derived from A549 whole cell lysates with α-phospho-serine (p-ser) antibody (top left panel) and α-PCBP1 antibody (bottom left panel) to examine TGF-β-dependent hnRNP E1 phosphorylation. The right panel depicts the quantification of band intensities analyzed by NIH Image J software. P-ser band intensity was normalized to the unstimulated 0 h condition. B IB analysis to confirm stable PCBP1 overexpression (PCBP1/OX) and silencing (PCBP1−/−) in A549 cells. C Role of PCBP1 on EMT is mediated by ILEI. IB analysis to analyze ILEI expression of whole cell lysates derived from A549 cells, A549 cells overexpressing PCBP1 (A549/PCBP1), and A549 cells with PCBP1 knockdown (A549/PCBP1 −/−)
ILEI could induce as well as maintain CD24lowCD44high subpopulation in A549 cells treated with TGF-β
It has been previously established that EMT is associated with enrichment of cell types with characteristic of stem cells (CD24lowCD44high) [20]. To further investigate whether ILEI played a potential role in induction or maintenance of CD24lowCD44high subpopulation of A549 cells treated with TGF-β, FACS analysis was carried out (Fig. 4). CD44 expression was significantly induced in A549 cells treated with TGF-β; median fluorescent intensity (MFI) was 129 ± 6 folds higher in the TGF-β treated A549 cells (Fig. 4A, left panel). To determine if ILEI expression can mimic the TGF-β-mediated induction of CD44 in the CD24low subpopulation of cells, we overexpressed ILEI (A549/ILEI) in A549 cells (data not shown). ILEI overexpression induced CD44 expression in untreated A549 cells in much the same way as TGF-β (Fig. 4A, middle panel); median fluorescent intensity (MFI) was 113 ± 17 folds higher in the ILEI overexpressing A549 cells. Conversely, stable knockdown of ILEI in A549 cells (data not shown) made them refractory to TGF-β-mediated induction of CD44 expression in the CD24low subpopulation of cells (Fig. 4A, right panel); MFI was 83 ± 9 folds lower in the ILEI knockdown A549 cells. In order to investigate if ILEI expression was required for enrichment of CD24lowCD44high subpopulation in A549 cells, we performed in vitro mammosphere assays with the parental A549 cells and both the ILEI overexpressing and ILEI silenced A549 cells. As shown in Fig. 4B, TGF-β treatment induced ability to form mammosphere in A549 cells. Just ILEI overexpression, without TGF-β treatment, induced more number of mammosphere compared to TGF-β treated parental A549 cells. Conversely, stable knockdown of ILEI completely inhibited mammosphere formation in A549 cells after TGF-β treatment (Fig. 4B). Taken together, these results showed that ILEI has a central role in both the induction and perhaps the maintenance of CD24lowCD44high subpopulation of TGF-β-mediated EMT in A549 cells.

ILEI induces and maintains the CD24lowCD44high subpopulation in A549 cells. A Shown are representative histograms of CD44 expression patterns in CD44highCD24low subpopulations of AA549 sorted by flow cytometry. At least 50,000 events were collected from each experiment. B The number of mammosphere formed in parental A549 cells or A549 cells, overexpressing ILEI (A549 cells/ILEI) or in which ILEI was stably silenced (A549 cells/ILEI−/−)
Discussion
It has been previously shown that A549 cells treated with TGF-β undergo EMT in a period of 3 days [21, 22], which is significantly more rapid than cell models transduced with transcription factors such as Snail, Slug, Twist, or Goosecoid [5]. However, in our study, distinct morphological changes associated with EMT was observed 24 h after TGF-β treatment in A549 cells. Downregulation of E-cadherin and upregulation of vimentin further demonstrated that 24 h of treatment with TGF-β could induce EMT of A549 cells.
ILEI, which was initially identified as a candidate gene for autosomal recessive nonsyndromic hearing loss locus 17 (DFNB17), was shown to be translationally upregulated during EMT in murine EpRas cells [12]. ILEI protein showed an increase after TGF-β treatment. However, when treated with the proteasomal inhibitor MG-132, ILEI protein levels did not show an induction in TGF-β-untreated A549 cells. This was consistent with the hypothesis that ILEI is translationally repressed in these cells. Hence, we successfully showed that ILEI expression is regulated at the post-transcriptional phase in human lung cancer cell line in much the same way as reported earlier in normal murine mammary gland and EpRas cells [7, 10, 12, 17].
Previous studies have shown that PCBP1 could bind to a structural, 33-nucleotide element in the 3′-UTR of ILEI that represses its translation in murine epithelial cells [7, 10, 17]. It was further shown that TGF-β activates Akt2, which in turn phosphorylates PCBP1 at serine-43. Phosphorylation of PCBP1 subsequently induced its release from the ILEI transcript and loss of translational silencing in TGF-β-treated cells [7, 10, 17]. In our study, a significant increase of PCBP1 phosphorylation was induced by TGF-β treatment. Moreover, overexpression of PCBP1 in A549 cells inhibited ILEI translation while constitutive expression of ILEI was shown in the A549/PCBP1−/− cells, even without TGF-β stimulation. Our result was supported by previous findings in murine cell line variants [7, 10, 17].
It has been widely proposed that both tumorigenesis and metastatic progression is contingent on transformation of a subpopulation of normal stem or progenitor cells, characterized by low expression of CD24 and high expression of CD44 [22–24].The inherent properties of stem/progenitor cells may impart their transformed counterparts with the ability to evade traditional antitumor therapies and to establish metastasis [23–25]. In the current study, TGF-β-treatment of A549 cells significantly induced CD44 expression. Overexpression of ILEI induced CD44 expression in much the same way as TGF-β. Nevertheless, stable knockdown of ILEI in A549 cells achieved a refractory result in the CD24low subpopulation of cells.
Cumulatively, these results clearly support our hypothesis that the role of PCBP1 in EMT and metastasis in lung cancer is mediated through induction of ILEI and that they are critical mediators of the aforementioned processes. Hence, we have functionally validated the existence of an evolutionary conserved transcript-selective translational regulation pathway by which TGF-β modulates expression of mRNA(s) required for EMT. However, it remains to be determined if this pathway is functioning to regulate other cognate transcripts required for EMT and whether it can be modulated to aid therapeutic intervention during metastatic lung cancer progression.
References
American Cancer Society. Global cancer facts and figures. 2nd ed. 2008. p15. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-027766.pdf. Accessed 10 Sep 2012.
World Health Organization. Cancer fact sheet. http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed 10 Sep 2012.
Van ZN. Neoadjuvant strategies for non-small cell lung cancer. Lung Cancer. 2001;34:s145–50.
Xiao DK, He JX. Epithelial mesenchymal transition and lung cancer. J Thorac Dis. 2010;2:154–9.
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumor progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28.
Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. 2010;12:286–93.
Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial–mesenchymal transition. Cancer Cell. 2009;15:402–15.
Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009;6:546–62.
Hussey GS, Chaudhury A, Dawson AE, Lindner DJ, Knudsen CR, Wilce MC, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41:419–31.
Petz M, Kozina D, Huber H, Siwiec T, Seipelt J, Sommergruber W, et al. The leader region of Laminin B1 mRNA confers cap-independent translation. Nucleic Acids Res. 2007;35:2473–82.
Waerner T, Alacakaptan M, Tamir I, Oberauer R, Gal A, Brabletz T, et al. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell. 2006;10:227–39.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tyskin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601.
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial–mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4.
Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907.
Wu X, Piper-Hunter MG, Crawford M, Nuovo GJ, Marsh CB, Otterson GA, et al. MicroRNAs in the pathogenesis of Lung Cancer. J Thorac Oncol. 2009;4:1028–34.
Hussey GS, Link LA, Brown AS, Howley BV, Chaudhury A, Howe PH. Establishment of a TGF-β-induced post-transcriptional EMT gene signature. PLoS ONE. 2012;7:e52624.
Wildey GM, Patil S, Howe PH. Smad3 potentiates transforming growth factor β (TGF-β)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J Biol Chem. 2003;278:18069–77.
Hocevar BA, Brown TL, Howe PH. TGFβ induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–56.
Mani SA, Guo W, Liao MJ, et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15.
Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–62.
Tauler J, Zudaire E, Liu H, Shih J, Mulshine JL. hnRNP A2/B1 modulates epithelial–mesenchymal transition in lung cancer cell lines. Cancer Res. 2010;70:7137–47.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Behbod F, Rosen JM. Will cancer stem cells provide new therapeutic targets? Carcinogenesis. 2005;26:703–11.
Dean M, Fojo T, Bates S. Tumor stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Qi Song and Wei Sheng are co-first authors.
Rights and permissions
About this article
Cite this article
Song, Q., Sheng, W., Zhang, X. et al. ILEI drives epithelial to mesenchymal transition and metastatic progression in the lung cancer cell line A549. Tumor Biol. 35, 1377–1382 (2014). https://doi.org/10.1007/s13277-013-1188-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-013-1188-y