
Abstract
The human gastrointestinal tract harbors a complex and abundant microbial community that can reach levels as high as 1013–1014 microorganisms in the colon. These microorganisms are essential to a host’s well-being in terms of nutrition and mucosa immunity. However, numerous studies have also implicated members of the colonic microbiota in the development of colorectal cancer (CRC). While CRC involves a genetic component where damaged DNA and genetic instability initiates a malignant transformation, environmental factors can also contribute to the onset of CRC. Furthermore, considering the constant exposure of the colonic mucosa to the microbiome and/or its metabolites, the mucosa has long been proposed to contribute to colon tumorigenesis. However, the mechanistic details of these associations remain unknown. Fortunately, due to technical and conceptual advances, progress in characterizing the taxonomic composition, metabolic capacity, and immunomodulatory activity of human gut microbiota have been made, thereby elucidating its role in human health and disease. Furthermore, the use of experimental animal models and clinical/epidemiological studies of environmental etiological factors has identified a correlation between gut microbiota composition and gastrointestinal cancers. Bacteria continuously stimulate activated immunity in the gut mucosa and also contribute to the metabolism of bile and food components. However, the highest levels of carcinogen production are also associated with gut anaerobic bacteria and can be lowered with live lactobacilli supplements. In this review, evidence regarding the relationship between microbiota and the development of CRC will be discussed, as well as the role for microbial manipulation in affecting disease development.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Within a malignant tumor, a complex community exists. This community can include oncogenically transformed cells with aberrant genomes, non-neoplastic cells including immune and stromal cells, and in some cases, microbes such as bacteria and viruses [1]. There are several types of cancer that are associated with infectious agents, and these cancers tend to occur in tissues that have a high level of exposure to microbes. Well-known examples include cervical cancer and gastric cancer, which can be caused by human papillomaviruses and the bacterium, Helicobacter pylori, respectively [2, 3].
For colon carcinogenesis, it is becoming increasingly evident that the large, complex bacterial population of the large intestine plays an important role [4]. For example, in animal models, mutant mice that are genetically susceptible to colorectal cancer (CRC) were found to develop significantly fewer tumors when maintained in germ-free conditions than in the presence of a conventional microbiota environment [5]. Therefore, it is possible that microbes influence multiple processes that affect cancer risk, including control of epithelial cell proliferation, differentiation, production of essential nutrients and/or bioactive food components, prevention of overgrowth of pathogenic organisms, inflammation, and stimulation of the intestinal immune system [4, 6, 7]. Furthermore, bacteria have been linked to CRC based on their production of toxic and genotoxic metabolites which can bind specific intestinal cell surface receptors and affect intracellular signal transduction. Carcinogenic agents may also be present in the diet or formed in vivo during digestion.
In this review, we will discuss the relationship between microbiota and the development of CRC which is emerging from experimental studies, as well as evidence that the microbial manipulation (probiotic) can impact disease development.
Overview of human gastrointestinal microbiota
Bacteria constitute approximately 90 % of all cells in the human body [8]. Furthermore, in the adult human gut, it is estimated that approximately 100 trillion microbial organisms reside, and these are collectively referred to as microbiota [9, 10]. Although the composition of the microbiota is remarkably stable at different anatomic locations along the gut, the absolute numbers of microbiota vary greatly [11]. For example, human gastrointestinal (GI) microbiota is present in the mouth, with 108–1010 colony-forming units (CFU) of bacteria present per gram saliva. With the swallowing reflex, these bacteria are constantly fed to the GI channel. Upon reaching the stomach, the number of bacteria is reduced to ~103 CFU/g gastric juice and then to 102–104 CFU/g content for the duodenum and jejunum. However, bacteria levels then increase in the ileum and colon to ~1010 and 1010–1012 CFU/g content, respectively [12]. The majority of these bacteria, estimated to constitute 1014 cells and more than 103 different species, colonize in the large intestine [13–15]. This complex ecosystem develops as a result of interactions between a host’s physiology and the bacteria that are introduced from the environment soon after birth [16]. In healthy adults, each person’s unique population of fecal microbiota is fairly stable over time, although fluctuations can occur in response to environmental, developmental, and pathological events [17]. Interestingly, the bacterial density in the large intestine (~1012 cells/ml) is much greater than in the small intestine (~102 cells/ml), and an approximately 12-fold higher risk of cancer is associated with the former than the latter [5].
Human intestinal microbiota has been found to be dominated by strict anaerobes including Bacteroides, Eubacterium, Bifidobacterium, Fusobacterium, Peptostreptococcus, and Atopobium [18]. In contrast, facultative anaerobes are present at approximately 1,000-fold lower levels and include Lactobacilli, Enterococci, Streptococci, and Enterobacteriaceae. There are more than 500 different bacterial species that may be present in normal commensal microbiota, although the exact number and variability among individuals remain to be characterized [19, 20]. In addition, specific strains of bacteria have been implicated in the pathogenesis of cancer, including Streptococcus bovis, Bacteroides, Clostridia, and H. pylori [21–24]. Conversely, some strains of bacteria, including Lactobacillus acidophilus and Bifidobacterium longum, have been shown to inhibit carcinogen-induced colon tumor development [25, 26]. Thus, it appears that the balance between “detrimental” and “beneficial” bacteria can affect the development of cancer. Correspondingly, a shift in the proportion of microbes present has been reported to influence carcinogen bioactivation and, thus, cancer risk.
Gut microbiota in the pathogenesis of colorectal cancer
It has long been thought that gut bacterial strains are inconsequential colonizers of the gut, while serving as pathogens when in the bowel. However, intestinal microbiota is now considered to have a symbiotic role with the host, which contributes to the maintenance of good health. Alternatively, though, gut bacteria can also be responsible for the development of chronic GI disorders when they are present at higher than normal concentrations or when they localize to regions of the gut that typically support low levels of bacteria. For example, Swidsinski and colleagues [27] detected high concentrations of bacteria present in 90 % of colonic biopsy specimens collected from patients with CRCs, in 93 % of specimens collected from patients with colonic adenomas, and in none of the specimens collected from asymptomatic controls. Therefore, bacterial or viral enteric infections may have a role in GI cancers [28]. Cuevas-Ramos and colleagues [29] also showed that adherent/invasive Escherichia coli strains were highly abundant in the colonic mucosa of patients with colorectal carcinoma and adenoma, yet not in normal colonic mucosa. These results demonstrate that a specific microorganism can be responsible for a malignant pathology. Similarly, S. bovis has been implicated in colonic neoplasia. Supplements of this strain of bacteria, as well as antigens extracted from the bacterial cell wall, have been shown to induce the formation of hyperproliferative aberrant colonic crypts and to increase the expression of proliferation markers in carcinogen-treated rats [30].
In 2011, three independent groups reported four high-resolution maps of human colonic dysbiosis associated with CRC [1, 31, 32]. All of these microbiome maps were generated from late-stage CRC tissue, and samples from both tumor tissues and surrounding nontumor tissues were examined. Similarities in the microbiome samples collected from the same individuals were found, despite the samples being from tumor and nontumor sites. A recent study also demonstrated that microbial communities of cancerous tissue and noncancerous tissues were similar based on an unweighted UniFrac principal component analysis (PCA) that was performed. Taken together, these results indicate that there is no marked difference in the microbial composition of tissues versus noncancerous tissues. However, the tumor microbiota did exhibit lower levels of diversity [33]. Despite the small sample size and differences in the experimental approaches used for these studies, it also appears that several bacterial species seem to preferentially inhabit either tumor or nontumor sites. The most striking similarity among the four documented CRC microbiomes was the enrichment of Fusobacterium spp. detected in the tumor samples. A histological analysis of a colorectal adenocarcinoma cell line also showed that these bacteria can invade tumor cells, thereby leading to their metastasis throughout the body. In addition, the relative abundance of Bacteroidaceae, Streptococcaceae, Fusobacteriaceae, Peptostreptococcaceae, Veillonellaceae, and Pasteurellaceae have been found to be significantly higher in cancerous tissues compared to the intestinal lumen. Furthermore, a significantly lower level of Lachnospiraceae, Ruminococcaceae, and Lactobacillaceae has been found in cancerous tissues compared to the intestinal lumen [33].
Conversely, nontumor sites appear to provide a niche for potentially pathogenic members of the family Enterobacteriaceae, such as Salmonella, Citrobacter, Cronobacter, and Shigella spp. [31], as well as members of the other Actinomycetales. Members of the phylum, Firmicutes, also exhibit a disparate distribution where some species are enriched in tumor tissues, while others inhabit the adjacent healthy mucosa of nontumor sites. These observations may reflect the ability of organisms that belong to the same taxonomic clade to mediate different functional roles in an ecosystem. Furthermore, this phenomenon may depend on the functional repertoire of the organisms involved, and these may include toxins, virulence factors, and other factors that facilitate interactions between bacteria and their environment [34].
Using a pyrosequencing-based analysis, Chen et al. examined 16S rRNA genes to profile the microbiota present in patients with CRC compared to healthy controls. Specifically, microbiota of the intestinal lumen, cancerous tissues, and matched noncancerous normal tissues were analyzed. In addition, mucosa-adherent microbes were examined from rectal swab samples, since tissue-adherent bacterial communities are potentially altered following bowel cleaning. The microbial structure of the intestinal lumen and cancerous tissues was found to differ considerably [33], with Firmicutes being more abundant and Bacteroidetes and Proteobacteria being less abundant in the lumen. Intestinal lumen microbiota and mucosa-adherent microbiota were also found to differ in CRC patients compared to matched healthy individuals. For example, the mucosa-adherent microbiota, Bifidobacterium, Faecalibacterium, and Blautia, were reduced in CRC patients, whereas Fusobacterium, Porphyromonas, Peptostreptococcus, and Mogibacterium were enriched. In the lumen, predominant phylotypes related to metabolic disorders or metabolic exchange with the host, Erysipelotrichaceae, Prevotellaceae, and Coriobacteriaceae were increased in CRC patients. These results suggest that the intestinal microbiota is associated with CRC risk and that intestinal lumen microflora potentially affects CRC risk primarily through direct interaction with the host. Gueimonde et al. [35] used quantitative reverse transcriptase polymerase chain reaction to analyze colonic mucosa samples obtained from 34 patients, including 21 patients with CRC, 9 patients with diverticulitis, and 4 patients with inflammatory bowel disease (IBD). For the patients with CRC, they had significantly lower levels of B. longum and Bifidobacterium bifidum compared to the other patients. Shen et al. [36] also evaluated adherent bacteria present in 21 adenoma and 23 non-adenoma subjects. After sequencing 335 clones for phylogenetic and taxonomic analyses, higher numbers of Proteobacteria and lower numbers of Bacteroidetes were detected in tumor subjects compared to control subjects. Furthermore, in an analysis of stool bacterial DNA using pyrosequencing and subsequent PCA, Sobhani et al. [37] detected a change in the composition of the microbiota of CRC patients. In particular, the Bacteroides/Prevotella species were found to be more numerous in cancer patients than in control subjects.
Taken together, these studies demonstrate that the gut microbiota may play an important role in CRC development, and this contribution can be detected quantitatively and qualitatively.
Possible mechanisms that link gut microbiota to the development of colorectal cancer
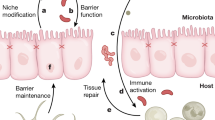
Gut microbiota reach their highest concentration in the large intestine, with over 1012 bacteria per gram of content [38]. Accordingly, the activities of this population have a significant impact on the health of the host. Therefore, if microbiota is involved in cancer development, the colon is predicted to be a major site of action. Animal models, including knockout mouse models and germ-free mice, have provided valuable insight into the role of bacteria in the development of CRC [39]. For example, the first observation linking the gut microbiota with CRC development was reported by Reddy et al. [40] in 1975. It was found that only 20 % of germ-free rats developed chemically induced CRC, while 93 % of conventionally maintained rats developed cancer and also exhibited multiple neoplasms. Since then, many animal models have shown that, under germ-free conditions, colitis and subsequent tumor formation are suppressed compared to either mono-associated or conventionalized animals [41–46]. Therefore, an important relationship between intestinal microbiota, potential effects on the immune system, and CRC has been established. Correspondingly, persistent immune dysregulation brought on by pathologic flora can contribute to inflammation and neoplastic changes in the mucosa. In addition, increased carcinogen production has been associated with intestinal anaerobic bacteria, as well as the conversion of primary bile acids to secondary bile salts, and these effects can contribute to the development of colon cancer [47]. Thus, intestinal microbiota appear to contribute to the development of CRC by inducing chronic inflammation and generating reactive metabolites and carcinogens [39] (Fig. 1).

An overview of the factors that contribute to colon cancer pathogenesis. Interactions between the microbiome, the immune balance of the colonic mucosa, and CECs, in addition to the genetics of CECs, are proposed to collectively contribute to the pathogenesis of colon cancer. In addition, a dynamic relationship exists among the GI microbiota, the intake and metabolism of food, and intestinal cells. For example, both the numbers and types of microbes and dietary factors can influence colon cancer risk and tumor behavior
The role of inflammation caused by an intestinal microbiota infection in the development of colorectal cancer
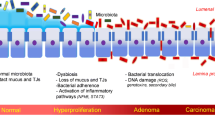
Chronic inflammation is associated with malignancy and has been proposed to be a contributing factor to a multitude of cancers [48]. In work by Kostic and coworkers [1], Fusobacterium sequences were found to be enriched in colorectal carcinomas based on quantitative polymerase chain reaction and a 16S rDNA sequence analysis performed for 95 carcinoma/normal DNA pairs. Fusobacteria were also visualized within colorectal tumors using fluorescence in situ hybridization. Based on these results, the Fusobacterium species may be associated with IBD, including both ulcerative colitis (UC) and Crohn’s disease. Furthermore, IBD is a known risk factor for CRC [49]. The link between inflammation and cancer was first suggested by Rudolf Virchow in 1863 when he noted the presence of leukocytes in neoplastic tissues. He hypothesized that this infiltrate mirrored the origin of cancer at sites with chronic inflammation [50]. Similarly, the concept that bacterial infection could promote tumorigenesis was first proposed in the late nineteenth century following the pioneering work of Robert Koch and Louis Pasteur that discovered bacteria at tumor sites [51]. Evidence for the involvement of microbiota in the induction of chronic colonic inflammation is growing, and it has become clear that chronic inflammation can profoundly alter local immune responses. For example, chronic inflammation can induce the release of reactive oxygen species and nitric oxide, which in turn can induce DNA damage and alter tissue homeostasis (Fig. 2). Consequently, it is hypothesized that a complex relationship exists between bacterial infection, inflammation, and tumorigenesis, and these interactions remain to be clarified. Moreover, cytokines and chemokines produced during inflammation can also act as tumor growth and survival factors and may induce tumor development by promoting angiogenesis and suppressing immune-mediated tumor elimination. Cancer-promoting cytokines include TNF-α, IL-6, and IL-1 among others, while mediators of inflammation also include TNF-α and IL-1, as well as IL-8, nitric oxide, prostaglandin-2 derivatives, and molecules of inflammatory pathway signaling. Both sets of factors have been shown to be involved in the progressive interplay that exists between immune cells and cells of a tissue undergoing transformation. Moreover, IL-10 and TGF-β have been shown to inhibit the incidence of CRC [52]. For example, Joshua et al. [53] demonstrated that conventionalized IL-10−/− mice exposed to the procarcinogenic compound, azoxymethane (AOM), developed spontaneous colitis and colorectal carcinomas, while AOM wild-type (WT) mice remained colitis-free and only developed low-grade dysplasia. Thus, the mechanisms by which bacterial agents may induce colonic carcinogenesis may include chronic inflammation and modulation of the immune reaction.

Intestinal microbiota promotes inflammation and carcinogenesis in the colon. Interactions between pattern recognition receptors (PRRs) and toll-like receptors (TLRs) with bacterial-associated molecular patterns trigger downstream signaling pathways that lead to the expression of various inflammatory mediators. Signaling from these mediators amplifies inflammation and promotes carcinogenesis in the colon. An additional consideration is the microbial enzymes of the metabolome that convert latent dietary procarcinogens into their biologically active forms and elicit neoplastic changes
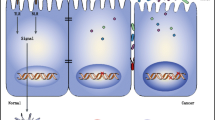
Several experimental models have been used to study the individual and collective roles of microorganisms during the development of inflammation and CRC. The normal microflora present in the gut is known to produce and release toxins which can bind specific cell surface receptors and affect intracellular signal transduction [54]. For example, although enterotoxigenic Bacteroides fragilis (ETBF) can asymptomatically colonize in humans, it can also secrete a B. fragilis toxin (BFT) which can cause human inflammatory diarrhea. Furthermore, this 20-kDa zinc-dependent metalloproteinase toxin has been shown to bind to colonic epithelial cells (CECs) and to stimulate cleavage of the tumor suppressor protein, E-cadherin [55] (Fig. 3). Additional mechanistic studies have identified that loss of membrane-associated E-cadherin in HT29/C1 cells triggers the nuclear localization of β-catenin, which then binds with T cell factor-dependent transcriptional activators to induce expression of c-Myc and cyclin D1. As a result, persistent cell proliferation is observed [56]. Activation of β-catenin signaling via mutations present in adenomatous polyposis coli complex proteins has also been found to contribute to the development of inherited and sporadic forms of CRC. Using multiple intestinal neoplasia (MIN) mice, Wu et al. [57] demonstrated that mice chronically colonized with ETBF developed colitis and colonic tumors. Although the exact mechanism by which bacteria affect the development of intestinal inflammation and cancer is not fully characterized, we hypothesize that the triggering of innate sensors, which are responsible for microbial detection, play an important role.

Inflammation-based initiation of colon carcinogenesis. STAT3/TH17 signaling provides a link between inflammation and carcinogenesis. When an innate signal triggers the inflammatory response, colonization and signaling downstream of NF-κB is activated. Production of proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 are also induced. ETBF colonization of CECs is accompanied by the production of BFT, which triggers the cleavage of E-cadherin and induces complex signal transduction in CECs involving the β-catenin/Wnt pathway. As a result, c-Myc is produced and CECs undergo proliferation and release cytokines/chemokines including IL-8 and TGFβ. Paracrine/autocrine expression of IL-6 and STAT3 activation may play central roles in promoting tumorigenesis via their pro-proliferative, antiapoptotic, and/or proangiogenesis properties. IL-6 secretion and STAT3 activation in epithelial and immune cells also contribute to diverting local T cell differentiation from a homeostatic regulatory pathway (T-reg) regulated by TGF-β to a Th17 proinflammatory response sustained by the production of key cytokines such as IL-23. IL-17 recruits polymorphonuclear leukocytes (PN) and may promote CEC proliferation through IL-6-dependent activation of STAT3. The respiratory burst originating from the antibacterial activity of PN is known to induce DNA damage and genetic instability, which can also initiate oncogenesis. Chronic asymptomatic ETBF colonization is proposed to result in a persistent Th17 inflammatory colonic response, which promotes CRC genesis, at least in part, through the actions of STAT3 and IL-6
The role of innate sensors in the development of colorectal cancer
The types and levels of activation of the various innate sensors can influence the gene expression pathways, as well as the level of inflammation. The consequences of these changes on DNA damage and chromatin alterations, which when combined with host genetic factors, can lead to tumorigenesis. Intestinal microbiota can elicit the cooperation of both innate and adaptive immune systems to protect a host and to maintain intestinal homeostasis [51, 58]. The innate immune system of a host has also been shown to play a role in regulating carcinogenesis. This regulation partly depends on specific pattern recognition receptors (PRRs) (Table 1), which are transmembrane or intracytoplasmic receptors that specifically recognize and bind highly conserved microbial signature molecules called pathogen-associated molecular patterns (PAMPs). These include lipopolysaccharide, flagellin, peptidoglycans, and formylated peptides among others.
PRRs are the most studied innate sensors relating to colitis and CRC and include the family of Toll-like receptors (TLRs). These receptors scan the extracellular space, while Nod-like receptors (NLRs) monitor the intracellular cytoplasmic compartment [7, 11]. TLRs were first described in Drosophila and are necessary for a host’s defense against Gram-positive bacteria. A similar system exists in plants where NLRs serve as the primary microbial sensors for plant immunity. Signaling pathways stimulated by PRRs are highly conserved. In the gut, the activation of PRRs initiates regulatory pathways including mitogen-activated protein kinase and nuclear factor κB (NF-κB)/Rel pathways, as well as caspase-dependent signaling cascades involving the inflammasome. To mediate interactions between gut microbiota and epithelial cells, a dynamic system is needed, and this is hypothesized to include NLRs and TLRs. Using a mouse model of colitis-associated cancer (CAC) induced with an injection of AOM or by repeated exposure to dextran sulfate sodium (DSS), Chen et al. [59] demonstrated that a greater number of tumors were established in Nod1-deficient mice than in WT mice. In addition, ApcMIN/+Nod−/− mice, which harbor a mutation in Apc and are also deficient in Nod1, developed a greater number of tumors as well. Based on these results, it appears that the Nod1 pathway enhances the tumor-promoting effect of attenuated Wnt signaling, which has been shown to play a critical role in colon tumorigenesis. Depletion of gut microbiota using antibiotic treatment has also been shown to suppress tumor development in Nod1-deficient mouse models.
TLRs belong to the TLR/interleukin-1 receptor (IL-1R) superfamily, and stimulation of TLRs by PAMPs can lead to the activation of NF-κB and the transcription of inflammatory cytokines and chemokine genes. However, it remains unclear how NF-κB-induced inflammation drives carcinogenesis. IL-6 induces the procarcinogenic signal transducer and activator of transcription (STAT3) pathway and also transcriptionally activates genes involved in cancer growth. Specifically, IL-6 activates proliferative, antiapoptotic, and proangiogenic genes such as c-IAP-1 and c-IAP-2, Fas ligand, c-Myc, p53, and cyclin D1. Accordingly, IL-6 is hypothesized to have a pivotal role in NF-κB-induced inflammation [60]. In addition, the single immunoglobulin IL-1 receptor-related molecule (SIGIRR), a negative regulator of Toll–IL-1R signaling, plays a critical role in gut homeostasis, intestinal inflammation, and colitis-associated tumorigenesis by maintaining the microbial tolerance of the colonic epithelium [61]. In human colonic samples, SIGIRR has been found to be expressed mainly in intraepidermal carcinomas and is expressed at significantly higher levels in inactive mucosa versus active mucosa [62]. In addition, colonic SIGIRR expression in mice has been observed to decrease rapidly following colitis development and then gradually return to basal levels [63]. It has been hypothesized that SIGIRR exerts its inhibitory effects by blocking the molecular interface of TLR4, TLR7, and the MyD88 adaptor mainly via its BB loop region [64]. Moreover, the innate adapter protein, MyD88, can prevent the development of CAC by transmitting IL-18 receptor signaling [65]. In contrast, TLR4 was found to promote the development of CRC in the AOM/DSS model [66]. TLR4 is expressed on CD4+ T cells, and when triggered, the phenotype of CD4+ T cells and their ability to provoke intestinal inflammation is affected. Taken together, these data support a role for the innate immune signaling pathways of the host in regulating inflammation-mediated colon cancer development. They also highlight the complex relationship that exists between microbiota, the inflammation response, and CRC in a host and support a model where susceptibility to developing CRC is modulated by microbiota and the repertoire of a host’s innate sensors.
ETBF: a representative microbiota in the carcinogenesis of colorectal cancer
Recently, deep sequencing technology has facilitated studies of microbial composition at both healthy and diseased body sites. Accordingly, Marchesi et al. [31] provide experimental data to support a possible role for intestinal microorganisms in CRC. On the basis of these data, a bacterial counterpart of the genetic driver–passenger model for CRC was developed [1, 31]. Briefly, bacterial drivers of CRC are defined as intestinal bacteria with procarcinogenic features that may initiate CRC development. In addition, the bacterial driver aspect of this model is related to the “alpha-bug” hypothesis that was recently proposed by Sears and colleagues [67]. This hypothesis suggests that alpha-bugs (such as EBTF) are directly pro-oncogenic and are capable of remodeling the mucosa immune response and colonic bacterial communities to further promote CRC (Fig. 4). One mechanism for this process involves the production of DNA-damaging compounds [68]. For example, Enterococcus faecalis is able to produce extracellular superoxide [69], and when this is converted to hydrogen peroxide, it has the potential to cause DNA damage in CECs [70, 71]. In addition, certain E. coli strains that harbor a polyketide synthetase island, which encodes a genotoxin called colibactin, can induce single-strand DNA breaks. Subsequent activation of DNA damage-induced signaling pathways then increases the mutation rate of infected cells [72].

Model of colon cancer induction by alpha-bugs. This model uses data for ETBF as an example of a putative alpha-bug. Alpha-bugs are directly pro-oncogenic and are also capable of remodeling the colonic bacterial community to enhance and further promote an induction of mucosal immune responses and CEC changes that result in colon cancer. Additionally, alpha-bugs may enhance carcinogenesis by selectively “crowding out” cancer-protective microbial species
ETBF strains have also been implicated in CRC initiation through the production of BFT (also known as fragilysin) [73, 74]. In addition to being directly genotoxic to CECs [75], this metalloproteinase stimulates cleavage of the tumor suppressor protein, E-cadherin, in intestinal epithelial cells [76]. The cleavage of E-cadherin then increases the permeability of the intestinal barrier and augments cell signaling via the β-catenin/Wnt pathway, which is constitutively active in most CRCs. Correspondingly, BFT is able to stimulate the proliferation and migration of human colon cancer cells in vitro [56]. The capacity for BFT to further activate the NF-κB pathway and induce the secretion of proinflammatory cytokines by CECs, combined with the observation that specific pools of NF-κB foster the initiation and promotion of epithelial tumorigenesis, has led to the hypothesis that ETBF are proinflammatory, oncogenic colonic bacteria. This hypothesis is also supported by a recent study conducted in Turkey which suggests that ETBF colonization is more frequent in CRC patients than in controls without CRC [74]. Furthermore, in a mouse model of ETBF-induced colitis and carcinogenesis [57], ETBF was found to enhance tumorigenesis by inducing the infiltration of the lamina propria by IL-17-producing CD4+ T cells (Th17) and γδ-T cells via STAT3 signaling (which is absolutely required for Th17 cell differentiation). This observation was made in both hyperplastic and adenomatous CECs, as well as in a subset of infiltrating immune cells. The role of IL-17 in ETBF tumorigenesis has also been demonstrated. For example, when ETBF-colonized MIN mice were treated with IL-17 blocking antibodies alone or in combination with antibodies targeting the receptor for IL-23 (the major cytokine that maintains Th17 cells [77, 78]), a decrease in the number of colonic tumors was observed. The role of IL-17 in tumorigenesis is further supported by recent data that show IL-17 can promote tumor growth in vitro and in vivo via the production of IL-6 by IL-17 receptor-bearing tumor cell lines [79]. Although the specific mechanism(s) involved have yet to be fully defined, it has been proposed that BFT induces a persistent TH17-type inflammatory response, and this may induce colonic epithelium carcinogenesis. This increase in IL-17 expression could also potentially activate STAT3 and IL-6, which have central roles in CRC as described previously [80, 81]. Furthermore, IL-17 has been shown to recruit polymorphonuclear leukocytes, and while the respiratory bursts from these cells are aimed at bacteria, they can also cause DNA damage and genetic instability in human cells, thereby potentially contributing to the initiation of CRC [80]. Overall, these results demonstrate that STAT3-dependent and TH17-dependent pathways have the potential to mediate inflammation-induced cancer by ETBF, thereby providing new insight into mechanisms relevant to human colon carcinogenesis.
Although definitive evidence is lacking, it is likely that members of the family Enterobacteriaceae also use a CRC-driving mechanism that is similar to the prolonged inflammatory response induced by ETBF. This family includes Shigella, Citrobacter, and Salmonella spp., and these, like ETBF, have been initially recognized as etiological agents of human diarrheal disease [82]. Accordingly, persistent low-grade colonization with such organisms could increase an individual’s susceptibility to CRC by inducing an asymptomatic, yet chronic, inflammatory response in the colonic mucosa [34]. Importantly, both NF-κB [83] and STAT-3 [84] are considered key mediators of inflammation-driven carcinogenesis via their putative antiapoptotic and cell cycle activity in CECs and their promotion of procarcinogenic mediators by immune cells. Therefore, we hypothesize that the composition of an individual’s colonic microbiota, repeated GI insults associated with life (e.g., ~1–2 episodes of diarrhea/person/year), and an individual’s proclivity to respond with a Th17 colonic mucosal response (which is likely to be dependent on genetics) determine an individual’s risk for developing critical CEC mutations that define colorectal carcinogenesis. Furthermore, ETBF is likely just one example of a microbiota organism expressing an oncogenic protease that can trigger a mucosa response that contributes to a malignant transformation. Additional support for this hypothesis is provided by the observation that polymorphisms in STAT-3 and the IL-23 receptor enhance the risk for chronic IBDs, which are inextricably connected with CRC risk.
The role of bacterial enzymes and metabolites in the development of colorectal cancer
The enormous number and diversity of human gut microbiota is reflected in a large and varied metabolic capacity, particularly in relation to xenobiotic biotransformation and carcinogen synthesis and activation. Moreover, these effects can have wide-ranging implications for the health of the host [30]. Based on interactions of the microflora with the host at both the local level and systemically, a broad range of immunological, physiological, and metabolic effects can be generated. Recently, a wide range of enzyme activities capable of generating potentially carcinogenic metabolites in the colon were proved to be associated with the gut microflora. These include β-glucuronidase, β-glucosidase, nitrate reductase, and nitroreductase enzymes [85, 86]. These enzymes are usually assayed in fecal suspensions and appear to be present in many types of bacteria [87–89]. They are also responsible for the conversion of inactive compounds to active metabolites, although this may involve adverse effects [90]. For example, bacterial transformation of dietary components and other chemicals in the intestinal lumen have been associated with the production of carcinogenic agents. As such, this process may represent another mechanism by which gut microbiota influence the development of CRC. Of these enzymes, β-glucuronidase and 7α-dehydroxylase have been the most extensively investigated as biomarkers of CRC risk
The metabolic activity of β-glucuronidase
Many carcinogenic compounds, as well as endogenously produced compounds such as steroids, are metabolized in the liver and then conjugated to glucuronic acid before being excreted via the bile into the small intestine. In the colon, bacteria β-glucuronidase hydrolyses the conjugates, thereby releasing the parent compound or its activated hepatic metabolite [4]. Metabolic epidemiological studies have shown that populations at high risk for CRC have high levels of fecal β-glucuronidase activity [88]. Furthermore, fecal β-glucuronidase activity in colon cancer patients has been found to be significantly higher than in healthy controls [88]. In the case of carcinogens and mutagens, β-glucuronidase activity in the colon may increase the likelihood of tumor induction. For example, the colon carcinogen, dimethylhydrazine (DMH), is metabolized in the liver, and small amounts of the procarcinogenic conjugate of the activated metabolite, methylazoxymethanol (MAM), are excreted in the bile. Hydrolysis of the conjugate by colonic bacteria releases MAM into the colon. For germ-free animals that have been treated with DMH, fewer colon tumors have been found to develop compared to conventional animals. In addition, when a β-glucuronidase inhibitor is administered in combination with the carcinogen, AOM, a significantly lower number of tumors in the rat colon were observed. This result indicates that microflora-derived β-glucuronidase plays an important role in the etiology of colon cancer [91] (Fig. 2). Furthermore, the activity of β-glucuronidase may be influenced by diet. For example, high-risk diets for CRC have consistently been shown to increase β-glucuronidase activity relative to low-risk diets [92]. Conversely, consumption of various types of fiber, including coffee fiber, resistant starch, and rice bran, have been associated with a decrease in β-glucuronidase activity in rats, with the extent of the effect dependent on the nature of the fiber [93]. In human studies, consumption of wheat bran, oat bran, and wholemeal rye have reduced β-glucuronidase activity [94]. A decrease in β-glucuronidase activity has also been associated with the use of B. longum as a dietary supplement in rats and humans. These results suggest that diet can influence the metabolic activity of certain types of intestinal microflora [95].
Bile acids and 7α-dehydroxylase
Bile acid consists of a number of related amphiphilic acidic steroids. Primary bile acids including chenodeoxycholic acid and cholic acid, which are synthesized from cholesterol in the liver, are then conjugated with taurine or glycine and released into the bile to solubilize fats and cholesterol for uptake in the small intestine. Intestinal microbiota play an important role in the metabolism of bile acids, predominantly involving the process of 7α-dehydroxylation, whereby cholic is converted to deoxycholic acid (DCA) and chenodeoxycholic is converted to lithocholic acid (LCA). This conversion increases the hydrophilicity of these secondary bile acids [96].
In an animal model, infusion of DCA was found to damage the mucosa of the intestinal tract, thereby inducing cell renewal. This was accompanied by an increase in cell proliferation, a process which may be a key mechanism in the effect of bile acids on colon carcinogenesis [48] (Fig. 5). Secondary bile acids have also been shown to induce DNA damage in colon cells, leading to apoptosis. DCA-induced DNA damage also triggers calcium ion-dependent apoptosis independent of p53 [97]. Moreover, the capacity for DCA to enhance colon tumor development in a rat model was shown be attenuated by all-trans retinoic acid [98]. Secondary bile acids may also influence CRC by supporting apoptosis-resistant cells or by mediating interactions with important secondary messenger signaling systems known to be activated in CRC [99].

Overview of the factors that derive from alterations in the microbial balance of the gut and contribute to CRC. ROI can cause DNA damage, and their numbers are higher during chronic inflammation and CRC, as observed in the fecal matrix. In addition, hydrophobic bile acids have been shown to promote colorectal carcinogenesis by inducing micronuclei formation, mitotic perturbations, and decreases in spindle checkpoint proteins
In a number of observational studies involving patients with adenomas or CRC, a correlation between fecal bile acid (FBA) concentrations and CRC risk has been observed [100]. For example, high fecal DCA concentrations and a high DCA-to-LCA ratio have been associated with an increased CRC risk [47]. However, not all studies have confirmed this relationship between bile acids and CRC risk [101]. For example, in epidemiological studies, concentrations of secondary bile acids were found to be higher in populations with a high risk for CRC. In addition, case–control studies demonstrated that 7α-dehydroxylase activity was higher in cases than in controls [91]. Based on our experience, high fat intake is another factor which correlates with CRC risk and increases FBA concentrations, while consumption of wheat bran reduces FBA concentrations. Furthermore, dietary manipulation resulting in a rise in colonic bile acid concentration has been shown to result in an increase in mucosa proliferation [102, 103]. Bile acids can disrupt the integrity of the cell membrane of colonic mucosal cells, leading to increased mucosa proliferation. Therefore, it has been hypothesized that hyperproliferation is induced by the cytotoxic potential of bile acids, and bile acids can directly stimulate or inhibit proliferation (Fig. 6). For example, bile salts can release prostaglandin E2 (PGE2) from colonic tissues, and the proliferative activity of CECs is suppressed by PGE2. Furthermore, bile salts can also enhance the release of arachidonate from colonocytes, which subsequently enhances the synthesis of PGE2 [104]. With secondary bile acids comprising over 80 % of FBAs and being able exert a range of biological and metabolic effects including cell necrosis, hyperplasia, tumor-promoting activity in the colon, induction of proliferation, DNA damage, and apoptosis [91, 105], it is predicted that the microbiota, as well as other factors, that modulate the levels and composition of bile acid play an important role in the etiology of colon cancer.

Role of diet and metabolites on colon carcinogenesis. Physiological considerations of ingesting meat, fiber lactulose, and calcium. The two main pathways that affect GI mucosa cells are meat-related mutagens and secondary bile acids. Mechanisms associated with the former remain to be determined. However, mechanisms associated with secondary bile acids can induce the formation of reactive oxygen and nitrogen species. This can lead to oxidative DNA damage and an increased risk of colon cancer associated with mutant cells
Genotoxicity of reactive oxygen intermediates
As mentioned earlier, bacteria have been linked to cancer via the induction of chronic inflammation following a bacterial infection and also by the production of toxic bacterial metabolites [106]. Bacteria can also bind potential mutagens, thereby reducing exposure of the host [107]. Animal studies have also shown that members of the commensal microbiota can produce metabolites that are potent direct-acting mutagens [108].
One such category of metabolites includes reactive oxygen intermediates (ROI). These molecules can generate oxidative DNA damage, and their numbers increase during chronic inflammation. Accumulating evidence also supports a role for ROI in the development of CRC, since ROI have been implicated in a wide range of cancers [109] (Fig. 5). ROI are derivatives of molecular oxygen and commonly include superoxides, hydrogen peroxide, hypochlorous acid, singlet oxygen, and hydroxyl radicals. They are produced in all cells during normal metabolism processes and can react with lipids and proteins to generate intermediates that react with DNA [110, 111]. However, independently, they can also cause alterations in DNA which include base modifications, deoxyribose damage, and breakage in DNA strands. These effects, coupled with a relatively slow and often incomplete repair process, can lead to chromosomal instability as a result of mutations, deletions, sister chromatid exchanges, and chromosomal translocations.
In a study by Huycke and colleagues, commensal E. faecalis was found to produce extracellular superoxide and hydrogen peroxide, and these molecules led to damage of CEC DNA both in vitro and in vivo [69]. For example, DNA damage was detected in the colon of rats colonized by either WT E. faecalis or a strain with attenuated extracellular superoxide production. Significantly more DNA damage was associated with the latter. Other studies have shown that this bacterium is capable of inducing IBD, dysplasia, and adenocarcinoma in IL-10 knockout mice, with an absence of pathology observed in germ-free mice [42]. Based on these experimental results, we hypothesize that ROI play an important role in the development of CRC by inducing damage to the DNA of epithelial cells of the colon during inflammation.
N-nitroso compounds
Nitrate, when ingested via diet and/or drinking water, is readily converted to nitrite, a more reactive and toxic product, by the nitrate reductase activity of the intestinal microflora. Nitrite is then available to react with nitrogenous compounds such as amines, amides, and methylureas in the body to produce NOC, many of which are highly carcinogenic, DNA-alkylating agents [112]. This reaction can occur in the acidic conditions that are prevalent in the human stomach or can be catalyzed at neutral pH by gut bacteria in the colon [113, 114]. The term, NOC, covers a wide range of compounds including N-nitrosamines, N-nitrosamides, N-nitrosoguanidines, and N-nitrosoureas, the majority of which are highly carcinogenic. Bacterial N-nitrosation can be analyzed using a method that determines apparent total NOC (ATNC) in feces and several biological fluids [4]. Such an approach has been used to demonstrate that N-nitrosation in the large intestine of rats is dependent on the presence of gut microflora. Although the mechanism responsible for bacterial N-nitrosation remains unknown, concentrations of ATNC have been found to positively correlate with intestinal transit time and to inversely correlate with fecal output. In addition, the ATNC excretion in fecal output increases with consumption of red meat, which has been associated with an elevated CRC risk in epidemiological studies.
Promotion of gastrointestinal health with probiotics
An individual’s diet can markedly influence the resident flora of the intestine. Accordingly, the use of diet to strategically alter the gut flora is a largely unexplored method to improve digestive and GI health. Probiotics represent an additional dietary consideration that has the potential to favorably alter the intestinal microbiota in order to prevent and treat GI diseases. Probiotic bacteria are often consumed in foods such as yogurts and cheese, in food supplements, or as drugs. Probiotics have been shown to favorably influence the development and stability of the intestinal microbiota, inhibit the colonization of pathogens, influence the mucosal barrier by trophic effects on the intestinal epithelium, protect against physiological stress, and stimulate both specific and nonspecific components of the immune system.
The term, probiotics, has been used for several decades and, in 2001, was defined as live microorganisms which confer a health benefit to the host when administered in adequate amounts [7]. Species of probiotics that are currently in use or under evaluation include Lactobacillus rhamnosus, Lactobacillus reuteri, L. acidophilus, Lactobacillus bulgaricus, Bifidobacterium infantis, Saccharomyces boulardii, Enterococcus faecium, the Nissle strain of E. coli, and Clostridium butyricum. Lactic acid bacteria and bifidobacteria are the most common types of microbes used as probiotics, while certain yeasts and bacilli may also be beneficial. In addition, probiotic microorganisms do not act exclusively in the large intestine to affect the intestinal flora. Probiotics can also affect other organs, either by modulating immunological parameters, intestinal permeability, and bacterial translocation or by providing bioactive metabolites, binding mutagens, and reducing inflammation [115, 116]. Other mechanisms by which probiotics positively affect the gut microbiota and liver health include an inhibition of intestinal bacterial enzymes, competition for limited nutrients, inhibition of bacteria mucosa adherence, and inhibition of epithelial cell invasion. At the molecular level, these activities can involve macrophage activation, a blocking of cytochrome P450, a reduction in carcinogen generation, downregulation of Ras-p21 expression, promotion of cell differentiation, inhibition of COX-2 upregulation, inhibition of nitric oxide synthase, an increase in short-chain fatty acid production, and a reduction in intestinal pH due to a reduction in the number of putrefactive bacteria [115, 117]. Probiotics can also meditate the delivery of anti-inflammatory mediators to downregulate proinflammatory cytokines, including INF-γ and TNF-α, via the NF-κB pathway.
Currently, a number of studies using a variety of probiotic strains have been conducted to determine the extent to which probiotics colonize and affect the GI tract. These studies have been reviewed by Cothesy et al. [118] and revealed that ingested strains do not become established members of the normal microbiota. Rather, probiotics may persist only during periods of dosing or for relatively short periods afterwards. Despite this, the effects of probiotic treatments in relation to cell proliferation and tumorigenesis have been observed. For example, Orlando et al. [119] found that administration of Lactobacillus GG induced a significant reduction in polyamine biosynthesis in both HGC-27 and DLD-1 cancer cell lines, thereby inducing an antiproliferative effect that was observed after 24 h. The antiproliferative capacity of probiotics may also relate to their ability to adhere to cells. Lee et al. [120] observed Bacillus polyfermenticus SCD to be strongly adherent to Caco-2 cells, and this probiotic was able to inhibit the growth of colon cancer cells in a dose-dependent manner. Kim et al. [121] also assessed the anticancer activity and bacterial enzyme inhibition of Bifidobacterium adolescentis SPM0212. This strain was able to inhibit the proliferation of three human colon cancer cell lines: HT-29, SW480, and Caco-2, and mediated a dose-dependent inhibition of TNF-α production that affected cell morphology. In addition, this particular bacterial strain was found to inhibit harmful fecal enzymes, including β-glucuronidase, β-glucosidase, tryptophanase, and urease. Furthermore, with daily oral administration of microencapsulated L. acidophilus, significant suppression of colon tumor incidence, tumor multiplicity, and reduced tumor size were observed [7]. More recently, enhanced apoptosis of carcinogen-damaged cells in rat colon was observed following the administration of both L. acidophilus and Bifidobacterium lactis with a resistant starch diet [122]. In contrast, the absence of a protective response was noted when a low resistant starch diet was consumed in the presence or absence of these two probiotics [123]. In rats treated with probiotics, fewer G1 intraepithelial neoplasias were observed, with the neoplasia present exhibiting a lower grade of dysplasia [7].
To date, well-controlled clinical studies to clearly document the therapeutic or preventive effects of probiotics in various diseases are scare. Even so, the therapeutic or preventive effects of certain probiotics have been documented in the therapy of pouchitis, traveler’s, and antibiotic-associated diarrhea, irritable bowel syndrome, and rotavirus enteritis [124]. In addition, consumption of probiotics by UC patients was found to prevent flare-ups and suppress the activation of NF-κB. Moreover, lower levels of TNF-α and IL-1β were detected, concomitant with an increase in IL-10 [12]. Additional clinical trials are needed to examine and verify the anti-inflammatory effects of probiotics in regulating systemic inflammation and local mucosa inflammation, as well as the ability of probiotics to regulate or correct immune-mediated diseases such as allergies and autoimmune diseases [12]. Optimization of dose and treatment period is also needed. However, probiotic treatments are expected to vary depending on the diversity of an individual’s microbiota, their genetic background, and the plasticity of their microbiota in response to diet. Nevertheless, based on data obtained from animal models, probiotics have been shown to mediate both local and systemic effects, to provide an inhibitory effect against pathogens, to optimize digestive processes, as well as to provide immunostimulative and antitumor activities.
Conclusion
It is becoming increasingly evident that microbiota in the large intestine play a key role in the etiology of CRC. Specifically, the presence of certain types of bacteria, as well as microbially generated metabolites, has been associated with the risk and development of CRC. With the details of these host–bacterial interactions becoming more apparent, the role of innate and adaptive immune responses is also being appreciated. In particular, the contribution of an inflammation state to the pathogenesis of CRC has been observed. Therefore, additional mechanistic studies are needed to further characterize the signaling pathways among intestinal bacteria, diet, and the immune system as it relates to the onset of CRC. In particular, the observation that probiotics can inhibit the inflammation process by enhancing the host immune response, by altering bacterial phylotypes in the colon, and by impacting the gut metabolome is of particular interest, especially since the administration of probiotics may provide a rational and standardized approach for the prevention and treatment of human CRC.
Abbreviations
- CRC:
-
Colorectal cancer
- PCA:
-
Principle component analysis
- IBD:
-
Inflammatory bowel diseases
- TGF-β:
-
Transforming growth factor-β
- ETBF:
-
Enterotoxigenic Bacteroides fragilis
- CECs:
-
Colonic epithelial cells
- BFT:
-
B. fragilis toxin
- PRRs:
-
Pattern recognition receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- TLRs:
-
Toll-like receptors
- NLRs:
-
Nod-like receptors
- NF-κB:
-
Nuclear factor κB
- DMH:
-
Dimethylhydrazine
- SIGIRR:
-
Single immunoglobulin IL-1 receptor-related molecule
- STAT3:
-
Signal transducer and activator of transcription
- MAM:
-
Metabolite methylazoxymethanol
- DCA:
-
Deoxycholic acid
- LCA:
-
Lithocholic acid
- FBA:
-
Fecal bile acid
- PGE2:
-
Prostaglandin E2
- NOC:
-
Nitroso compounds
- IFN-γ:
-
Interferon-γ
- TNF-α:
-
Tumor necrosis factor-α
- CFU:
-
Colony-forming units
- GI:
-
Gastrointestinal
- ROI:
-
Reactive oxygen intermediate
- H2S:
-
Hydrogen sulfide
References
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22(2):292–8.
Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392(1):1–10.
Warren JR. Helicobacter: the ease and difficulty of a new discovery. Chem Med Chem. 2006;1(7):672–85.
Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des. 2009;15(13):1524–7.
Proctor LM. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe. 2011;10(4):287–91.
Greer JB, O'Keefe SJ. Microbial induction of immunity, inflammation, and cancer. Front Physiol. 2011;1:168.
Zhu Y, Michelle Luo T, Jobin C, Young HA. Gut microbiota and probiotics in colon tumorigenesis. Cancer Lett. 2011;309(2):119–27.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65.
Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–33.
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65(11):4799–807.
Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136(1):65–80.
Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3(6):637–82.
Dethlefsen L, Eckburg PB, Bik EM, Relman DA. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21(9):517–23.
Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4586–91.
Marchesi JR. Human distal gut microbiome. Environ Microbiol. 2011;13(12):3088–102.
Goncharova GI, Dorofeĭchuk VG, Smolianskaia AZ, Sokolova KIA. Microbial ecology of the intestines in health and in pathology. Antibiot Khimioter. 1989;34(6):462–6.
Stanghellini V, Barbara G, Cremon C, Cogliandro R, Antonucci A, Gabusi V, et al. Gut microbiota and related diseases: clinical features. Intern Emerg Med. 2010;5 Suppl 1:S57–63.
Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol Lett. 2004;93(2–3):97–108.
Rastall RA. Bacteria in the gut: friends and foes and how to alter the balance. J Nutr. 2004;134(8 Suppl):2022S–6S.
Mai V. Dietary modification of the intestinal microbiota. Nutr Rev. 2004;62(6 Pt 1):235–42.
Peek Jr RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2(1):28–37.
Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy. Arch Surg. 2004;139(7):760–5.
Moore WE, Moore LH. Intestinal floras of populations that have a high risk of colon cancer. Appl Environ Microbiol. 1995;61(9):3202–7.
Nakamura J, Kubota Y, Miyaoka M, Saitoh T, Mizuno F, Benno Y. Comparison of four microbial enzymes in Clostridia and Bacteroides isolated from human feces. Microbiol Immunol. 2002;46(7):487–90.
McIntosh GH, Royle PJ, Playne MJ. A probiotic strain of L. acidophilus reduces DMH-induced large intestinal tumors in male Sprague-Dawley rats. Nutr Cancer. 1999;35(2):153–9.
Rowland IR, Bearne CA, Fischer R, Pool-Zobel BL. The effect of lactulose on DNA damage induced by DMH in the colon of human flora-associated rats. Nutr Cancer. 1996;26(1):37–47.
Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, et al. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115(2):281–6.
de Martel C, Franceschi S. Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol. 2009;70(3):183–94.
Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107(25):11537–42.
Ellmerich S, Djouder N, Schöller M, Klein JP. Production of cytokines by monocytes, epithelial and endothelial cells activated by Streptococcus bovis. Cytokine. 2000;12(1):26–31.
Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, et al. Towards the human colorectal cancer microbiome. PLoS One. 2011;6(5):e20447.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306.
Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7(6):e39743.
Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver–passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–82.
Gueimonde M, Ouwehand A, Huhtinen H, Salminen E, Salminen S. Qualitative and quantitative analyses of the bifidobacterial microbiota in the colonic mucosa of patients with colorectal cancer, diverticulitis and inflammatory bowel disease. World J Gastroenterol. 2007;13(29):3985–9.
Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1(3):138–47.
Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6(1):e16393.
Cummings JH, Macfarlane GT. The control and consequences of bacterial fermentation in the human colon. J Appl Bacteriol. 1991;70(6):443–59.
Hope ME, Hold GL, Kain R, El-Omar EM. Sporadic colorectal cancer—role of the commensal microbiota. FEMS Microbiol Lett. 2005;244(1):1–7.
Reddy BS, Mastromarino A, Wynder EL. Further leads on metabolic epidemiology of large bowel cancer. Cancer Res. 1975;35(11 Pt. 2):3403–6.
Marteau PR, de Vrese M, Cellier CJ, Schrezenmeir J. Protection from gastrointestinal diseases with the use of probiotics. Am J Clin Nutr. 2001;73(2 Suppl):430S–6S.
Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. 2002;160(6):2253–7.
Kado S, Uchida K, Funabashi H, Iwata S, Nagata Y, Ando M, et al. Intestinal microflora are necessary for development of spontaneous adenocarcinoma of the large intestine in T-cell receptor beta chain and p53 double-knockout mice. Cancer Res. 2001;61(6):2395–8.
Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92(5):645–56.
Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E, et al. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002;62(22):6362–6.
Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003;162(2):691–702.
Owen RW. Faecal steroids and colorectal carcinogenesis. Scand J Gastroenterol Suppl. 1997;222:76–82.
Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front Immunol. 2012;3:107.
Moossavi S, Bishehsari F. Inflammation in sporadic colorectal cancer. Arch Iran Med. 2012;15(3):166–70.
Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI—atheromatous affection of arteries. 1858. Nutr Rev. 1989;47(1):23–5.
Compare D, Nardone G. Contribution of gut microbiota to colonic and extracolonic cancer development. Dig Dis. 2011;29(6):554–61.
Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101.e5–14.e5.
Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4(6):e6026.
Fasano A. Cellular microbiology: can we learn cell physiology from microorganisms? Am J Physiol. 1999;276(4 Pt 1):C765–76.
Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77(4):1708–18.
Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124(2):392–400.
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–22.
DuPont AW, DuPont HL. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol. 2011;8(9):523–31.
Chen GY, Shaw MH, Redondo G, Núñez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68(24):10060–7.
Zhang G, Ghosh S. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107(1):13–9.
Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4(9):920–7.
Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26(4):461–75.
Kadota C, Ishihara S, Aziz MM, Rumi MA, Oshima N, Mishima Y, et al. Down-regulation of single immunoglobulin interleukin-1R-related molecule (SIGIRR)/TIR8 expression in intestinal epithelial cells during inflammation. Clin Exp Immunol. 2010;162(2):348–61.
Gong J, Wei T, Stark RW, Jamitzky F, Heckl WM, Anders HJ, et al. Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: a computational approach. J Struct Biol. 2010;169(3):323–30.
Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207(8):1625–36.
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–81.
Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis. 2011;203(3):306–11.
Boleij A, Tjalsma H. Gut bacteria in health and disease: a survey on the interface between intestinal microbiology and colorectal cancer. Biol Rev Camb Philos Soc. 2012;87(3):701–30.
Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002;23(3):529–36.
Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology. 2007;132(2):551–61.
Wang X, Allen TD, May RJ, Lightfoot S, Houchen CW, Huycke MM. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 2008;68(23):9909–17.
Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–51.
Wu S, Shin J, Zhang G, Cohen M, Franco A, Sears CL. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect Immun. 2006;74(9):5382–90.
Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T, et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect. 2006;12(8):782–6.
Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011;108(37):15354–9.
Wu S, Rhee KJ, Zhang M, Franco A, Sears CL. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma-secretase-dependent E-cadherin cleavage. J Cell Sci. 2007;120(Pt 11):1944–52.
McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204(7):479–90.
Näthke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–66.
Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–64.
Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/−) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9(1):3–5.
Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4.
DuPont HL. Clinical practice. Bacterial diarrhea. N Engl J Med. 2009;361(16):1560–9.
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6.
Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51.
Cole CB, Fuller R, Mallet AK, Rowland IR. The influence of the host on expression of intestinal microbial enzyme activities involved in metabolism of foreign compounds. J Appl Bacteriol. 1985;59(6):549–53.
Steer TE, Johnson IT, Gee JM, Gibson GR. Metabolism of the soybean isoflavone glycoside genistin in vitro by human gut bacteria and the effect of prebiotics. Br J Nutr. 2003;90(3):635–42.
Takada H, Hirooka T, Hiramatsu Y, Yamamoto M. Effect of beta-glucuronidase inhibitor on azoxymethane-induced colonic carcinogenesis in rats. Cancer Res. 1982;42(1):331–34.
Kim DH, Jin YH. Intestinal bacterial beta-glucuronidase activity of patients with colon cancer. Arch Pharm Res. 2001;24(6):564–7.
Rowland IR, Tanaka R. The effects of transgalactosylated oligosaccharides on gut flora metabolism in rats associated with a human faecal microflora. J Appl Bacteriol. 1993;74(6):667–74.
Davis CD, Milner JA. Gastrointestinal microflora, food components and colon cancer prevention. J Nutr Biochem. 2009;20(10):743–52.
Gill CI, Rowland IR. Diet and cancer: assessing the risk. Br J Nutr. 2002;88 Suppl 1:S73–87.
Reddy BS, Mangat S, Weisburger JH, Wynder EL. Effect of high-risk diets for colon carcinogenesis on intestinal mucosal and bacterial beta-glucuronidase activity in F344 rats. Cancer Res. 1977;37(10):3533–6.
Mazière S, Meflah K, Tavan E, Champ M, Narbonne JF, Cassand P. Effect of resistant starch and/or fat-soluble vitamins A and E on the initiation stage of aberrant crypts in rat colon. Nutr Cancer. 1998;31(3):168–77.
Gråsten SM, Juntunen KS, Poutanen KS, Gylling HK, Miettinen TA, Mykkänen HM. Rye bread improves bowel function and decreases the concentrations of some compounds that are putative colon cancer risk markers in middle-aged women and men. J Nutr. 2000;130(9):2215–21.
Lee JW, Shin JG, Kim EH, Kang HE, Yim IB, Kim JY, et al. Immunomodulatory and antitumor effects in vivo by the cytoplasmic fraction of Lactobacillus casei and Bifidobacterium longum. J Vet Sci. 2004;5(1):41–8.
de Giorgio R, Blandizzi C. Targeting enteric neuroplasticity: diet and bugs as new key factors. Gastroenterology. 2010;138(5):1663–6.
Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol. 2001;33(2):193–203.
Narahara H, Tatsuta M, Iishi H, Baba M, Uedo N, Sakai N, et al. K-ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acid. Int J Cancer. 2000;88(2):157–61.
Radley S, Davis AE, Imray CH, Barker G, Morton DG, Baker PR, et al. Biliary bile acid profiles in familial adenomatous polyposis. Br J Surg. 1992;79(1):89–90.
Imray CH, Radley S, Davis A, Barker G, Hendrickse CW, Donovan IA, et al. Faecal unconjugated bile acids in patients with colorectal cancer or polyps. Gut. 1992;33(9):1239–45.
de Kok TM, van Maanen JM. Evaluation of fecal mutagenicity and colorectal cancer risk. Mutat Res. 2000;463(1):53–101.
Deschner EE, Cohen BI, Raicht RF. Acute and chronic effect of dietary cholic acid on colonic epithelial cell proliferation. Digestion. 1981;21(6):290–6.
Deschner EE, Raicht RF. Influence of bile on kinetic behavior of colonic epithelial cells of the rat. Digestion. 1979;19(5):322–7.
DeRubertis FR, Craven PA, Saito R. Bile salt stimulation of colonic epithelial proliferation. Evidence for involvement of lipoxygenase products. J Clin Invest. 1984;74(5):1614–24.
Hughes R, Kurth MJ, McGilligan V, McGlynn H, Rowland I. Effect of colonic bacterial metabolites on Caco-2 cell paracellular permeability in vitro. Nutr Cancer. 2008;60(2):259–66.
Heavey PM, Rowland IR. Microbial–gut interactions in health and disease. Gastrointestinal cancer. Best Pract Res Clin Gastroenterol. 2004;18(2):323–36.
Orrhage K, Sillerström E, Gustafsson JA, Nord CE, Rafter J. Binding of mutagenic heterocyclic amines by intestinal and lactic acid bacteria. Mutat Res. 1994;311(2):239–48.
Carman RJ, Van Tassell RL, Kingston DG, Bashir M, Wilkins TD. Conversion of IQ, a dietary pyrolysis carcinogen to a direct-acting mutagen by normal intestinal bacteria of humans. Mutat Res. 1988;206(3):335–42.
Owen RW, Spiegelhalder B, Bartsch H. Generation of reactive oxygen species by the faecal matrix. Gut. 2000;46(2):225–32.
Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21(3):361–70.
Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park). 2002; 16(2):217–26, 229.
Hughes R, Cross AJ, Pollock JR, Bingham S. Dose-dependent effect of dietary meat on endogenous colonic N-nitrosation. Carcinogenesis. 2001;22(1):199–202.
Massey RC, Key PE, Mallett AK, Rowland IR. An investigation of the endogenous formation of apparent total N-nitroso compounds in conventional microflora and germ-free rats. Food Chem Toxicol. 1988;26(7):595–600.
Rowland IR, Granli T, Bøckman OC, Key PE, Massey RC. Endogenous N-nitrosation in man assessed by measurement of apparent total N-nitroso compounds in faeces. Carcinogenesis. 1991;12(8):1395–401.
Pala V, Sieri S, Berrino F, Vineis P, Sacerdote C, Palli D, et al. Yogurt consumption and risk of colorectal cancer in the Italian European prospective investigation into cancer and nutrition cohort. Int J Cancer. 2011;129(11):2712–9.
de Vrese M, Schrezenmeir J. Probiotics, prebiotics, and synbiotics. Adv Biochem Eng Biotechnol. 2008;111:1–66.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80.
Corthésy B, Gaskins HR, Mercenier A. Cross-talk between probiotic bacteria and the host immune system. J Nutr. 2007;137(3 Suppl 2):781S–90S.
Orlando A, Messa C, Linsalata M, Cavallini A, Russo F. Effects of Lactobacillus rhamnosus GG on proliferation and polyamine metabolism in HGC-27 human gastric and DLD-1 colonic cancer cell lines. Immunopharmacol Immunotoxicol. 2009;31(1):108–16.
Lee NK, Park JS, Park E, Paik HD. Adherence and anticarcinogenic effects of Bacillus polyfermenticus SCD in the large intestine. Lett Appl Microbiol. 2007;44(3):274–8.
Kim Y, Lee D, Kim D, Cho J, Yang J, Chung M, et al. Inhibition of proliferation in colon cancer cell lines and harmful enzyme activity of colon bacteria by Bifidobacterium adolescentis SPM0212. Arch Pharm Res. 2008;31(4):468–73.
Pool-Zobel BL, Neudecker C, Domizlaff I, Ji S, Schillinger U, Rumney C, et al. Lactobacillus- and Bifidobacterium-mediated antigenotoxicity in the colon of rats. Nutr Cancer. 1996;26(3):365–80.
Le Leu RK, Brown IL, Hu Y, Bird AR, Jackson M, Esterman A, et al. A synbiotic combination of resistant starch and Bifidobacterium lactis facilitates apoptotic deletion of carcinogen-damaged cells in rat colon. J Nutr. 2005;135(5):996–1001.
Tlaskalová-Hogenová H, Stěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8(2):110–20.
Acknowledgments
We thank Dr. Huanlong Qin for his critical reading of this manuscript.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhu, Q., Gao, R., Wu, W. et al. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumor Biol. 34, 1285–1300 (2013). https://doi.org/10.1007/s13277-013-0684-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-013-0684-4