
Abstract
Aberrant DNA methylation has been implicated in the development of hepatocellular carcinoma (HCC). Our aim was to clarify its molecular mechanism and to identify useful biomarkers by screening for DNA methylation in HCC. Methylated CpG island amplification coupled with CpG island microarray (MCAM) analysis was carried out to screen for methylated genes in primary HCC specimens [hepatitis B virus (HBV)-positive, n = 4; hepatitis C virus (HCV)-positive, n = 5; HBV/HCV-negative, n = 7]. Bisulfite pyrosequencing was used to analyze the methylation of selected genes and long interspersed nuclear element (LINE)-1 in HCC tissue (n = 57) and noncancerous liver tissue (n = 50) from HCC patients and in HCC cell lines (n = 10). MCAM analysis identified 332, 342, and 259 genes that were methylated in HBV-positive, HCV-positive, and HBV/HCV-negative HCC tissues, respectively. Among these genes, methylation of KLHL35, PAX5, PENK, and SPDYA was significantly higher in HCC tissue than in noncancerous liver tissue, irrespective of the hepatitis virus status. LINE-1 hypomethylation was also prevalent in HCC and correlated positively with KLHL35 and SPDYA methylation. Receiver operating characteristic curve analysis revealed that methylation of the four genes and LINE-1 strongly discriminated between HCC tissue and noncancerous liver tissue. Our data suggest that aberrant hyper- and hypomethylation may contribute to a common pathogenesis mechanism in HCC. Hypermethylation of KLHL35, PAX, PENK, and SDPYA and hypomethylation of LINE-1 could be useful biomarkers for the detection of HCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common human malignancies, worldwide [1]. Chronic infection by hepatitis B virus (HBV) and hepatitis C virus (HCV) are well-documented risk factors for the development of HCC, while chronic alcoholism and various environmental factors, including aflatoxin B1, are also believed to be important risk factors [2, 3]. The development and progression of HCC is often a complex, multistep process entailing the evolution of normal liver through chronic hepatitis and cirrhosis to HCC, but HCC can also arise in a noncirrhotic liver. In either case, the process is influenced by multiple genetic changes, including allelic deletions, chromosomal losses and gains, DNA rearrangements, and gene mutations [4]. In addition, a growing body of evidence suggests that epigenetic changes such as DNA methylation and histone modification also play crucial roles in hepatocarcinogenesis.
Two seemingly contradictory epigenetic events coexist in cancer: global hypomethylation, which is mainly observed in repetitive sequences throughout the genome, and regional hypermethylation, which is frequently associated with CpG islands within gene promoters [5]. Hypermethylation of CpG islands is a common feature of cancer and is associated with gene silencing. Although the classical two-hit theory posits that tumor suppressor genes are inactivated by gene mutation or deletion, it is now recognized that DNA hypermethylation is a third mechanism by which inactivation of tumor suppressor genes occurs, and that it plays a significant role in tumorigenesis. In contrast to the CpG islands, repetitive DNA elements are normally heavily methylated in somatic tissues. About 45 % of the human genome is composed of repetitive sequences, including long interspersed nuclear elements (LINEs) and short interspersed nuclear element [6], and studies have shown that methylation of such repetitive elements can serve as a surrogate for the global methylcytosine content [7]. In that regard, LINE-1 hypomethylation is known to occur during the development of various human malignancies, including HCC [8, 9].
HCC is generally diagnosed at an advanced stage of tumor progression, and a large fraction of HCC cases are fatal. Thus, a better understanding of the underlying molecular mechanisms and identification of genes critical for early detection of HCC and therapeutic intervention would be highly desirable. Although a number of hyper- or hypomethylated loci have been identified in HCC [10–12], only a few studies have been conducted to unravel the genome-wide methylation status [13–15]. In the present study, we carried out genome-wide CpG island methylation analysis in a set of primary HCC specimens, with and without hepatitis virus infection. We also evaluated the hypomethylation of LINE-1 and assessed its association with aberrant CpG island hypermethylation in HCC.
Materials and methods
Tissue samples and cell lines
A total of 57 primary HCC specimens (HBV-positive, n = 21; HCV-positive, n = 21; HBV/HCV-negative, n = 15) were obtained through surgical resection or needle biopsy at Sapporo Medical University Hospital. Corresponding samples of noncancerous liver tissue were also obtained from 50 patients. HBV surface (HBs) antigen and anti-HCV antibody were measured serologically. An informed consent was obtained from all patients before collection of the specimens. The ten liver cancer cell lines (HT17, PLC/PRF/5, Li-7, huH-1, HuH-7, HepG2, Hep3B, HLE, HLF, and JHH-4) used have been described previously [11]. To analyze restoration of gene expression, cells were treated with 2.0 μM 5-aza-2′-deoxycytidine (5-aza-dC) (Sigma, St Louis, MO, USA) for 72 h, replacing the drug and medium every 24 h. Genomic DNA was extracted using the standard phenol–chloroform procedure. Total RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) and then treated with a DNA-free kit (Ambion, Austin, TX, USA). Genomic DNA and total RNA from normal liver tissue from a healthy individual were purchased from BioChain (Hayward, CA, USA).
Methylated CpG island amplification coupled with CpG island microarray
Methylated CpG island amplification (MCA) was performed as described previously [13]. Briefly, 500 ng of genomic DNA was digested with the methylation-sensitive restriction endonuclease SmaI (New England Biolabs, Ipswich, MA, USA), after which it was digested with the methylation-insensitive restriction endonuclease XmaI. The adaptors were prepared by addition of the oligonucleotides RMCA12 (5′-CCGGGCAGAAAG-3′) and RMCA24 (5′-CCACCGCCATCCGAGCCTTTCTGC-3′). After ligation of the digested DNA to the adaptors, PCR amplification was carried out. Using a BioPrime Plus Array CGH Genomic Labeling System (Invitrogen), MCA amplicons from the HCC samples were labeled with Alexa Fluor 647, while amplicons from a normal liver sample was labeled with Alexa Fluor 555. The labeled MCA amplicons were then hybridized to a custom human CpG island microarray containing 15,134 probes covering 6,157 unique genes (G4497A; Agilent Technologies, Santa Clara, CA, USA) [16]. After washing, the array was scanned using an Agilent DNA Microarray Scanner (Agilent technologies), and the data were processed using Feature Extraction software ver. 10.7 (Agilent Technologies). The data were then analyzed using GeneSpring GX ver. 11 (Agilent Technologies).
Methylation-specific PCR
Genomic DNA (1 μg) was modified with sodium bisulfite using an EpiTect Bisulfite Kit (Qiagen, Hilden, Germany), and methylation-specific PCR (MSP) was performed as described previously [17]. Briefly, PCR was run in a 25-μl volume containing 50 ng of bisulfite-treated DNA, 1× MSP buffer [67 mM Tris–HCl (pH 8.8), 16.6 mM (NH4)2SO4, 6.7 mM MgCl2, and 10 mM 2-mercaptoethanol], 1.25 mM dNTP, 0.4 μM each primer, and 0.5 U of JumpStart REDTaq DNA Polymerase (Sigma). The PCR protocol for MSP entailed 5 min at 95°C; 35 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C; and a 7 min final extension at 72°C. Primer sequences and PCR product sizes are shown in Supplementary Table 1.
Bisulfite pyrosequencing analysis
Bisulfite pyrosequencing analysis was performed as described previously [17]. The PCR protocol entailed 5 min at 95°C; 45 cycles of 1 min at 95°C, 1 min at 60°C, and 1 min at 72°C; and a 7-min final extension at 72°C. PCR products were then bound to Streptavidin Sepharose beads HP (Amersham Biosciences, Piscataway, NJ); after which, the beads containing the immobilized PCR product were purified, washed, and denatured using a 0.2 M NaOH solution. After addition of 0.3 μM sequencing primer to the purified PCR product, pyrosequencing was carried out using a PSQ96MA system (Qiagen, Hilden, Germany) and Pyro Q-CpG software (Qiagen). Primer sequences and PCR product sizes are shown in Supplementary Table 1.
Quantitative RT-PCR
Single-stranded cDNA was prepared using SuperScript III reverse transcriptase (Invitrogen). Quantitative RT-PCR was carried out using TaqMan Gene Expression Assays (KLHL35, Hs00400533_m1; PAX5, Hs00172003_m1; PENK, Hs00175049_m1; SPDYA, Hs00736925_m1; GAPDH, Hs99999905_m1; Applied Biosystems, Foster City, CA, USA) and a 7500 Fast Real-Time PCR System (Applied Biosystems) according to the manufacturer's instructions. SDS1.4 software (Applied Biosystems) was used for comparative delta Ct analysis, and GAPDH served as an endogenous control.
Statistical analysis
To compare differences in continuous variables between groups, t tests or ANOVA with post hoc Tukey's tests were performed. Fisher's exact test or chi-squared test was used for analysis of categorical data. Receiver operator characteristic (ROC) curves were constructed based on the levels of methylation. Values of P < 0.05 (two-sided) were considered statistically significant. Statistical analyses were carried out using SPSS statistics 18 (IBM Corporation, Somers, NY, USA) and GraphPad Prism ver. 5.0.2 (GraphPad Software, La Jolla, CA, USA).
Results
Genome-wide CpG island methylation analysis in HCC
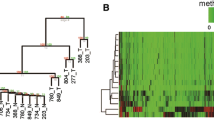
To screen for CpG island hypermethylation in HCC, we carried out methylated CpG island amplification coupled with CpG island microarray (MCAM) analysis using a set of HCC tissue specimens (HBV-positive, n = 4; HCV-positive, n = 5; HBV/HCV-negative, n = 7). As in an earlier study in which the same array system was used, we utilized a signal ratio (Cy5/Cy3) of >2.0 as the criterion for a methylation-positive probe [13]. The average number of methylated probe sets in the HCC specimens was 566 (range 159–846). To assess the association between hepatitis virus infection and methylation status, we categorized the HCC specimens according to their viral status. The average numbers of methylated probe sets in HBV-positive, HCV-positive, and the HBV/HCV-negative HCC specimens were 574, 598, and 539, respectively, which did not significantly differ (P = 0.840). Interestingly, however, the numbers of methylated probe sets were more varied among HBV/HCV-negative HCCs, which is indicative of their varied pathological backgrounds (Fig. 1a).

Genome-wide analysis of CpG island methylation. a MCAM analysis was carried out using a series of HCC tissue specimens (HBV-positive, n = 4; HCV-positive, n = 5; HBV/HCV-negative, NBNC, n = 7). MCAM data were categorized into three groups based on the hepatitis virus status, and the numbers of methylated genes in the respective categories are shown. b Venn diagram analysis of the methylated genes in the indicated categories. c Gene tree view of the MCAM analysis results. A set of 714 probes (514 unique genes) were selected as commonly methylated genes, after which, hierarchical clustering was performed. Each row represents a single probe
To identify commonly methylated genes in HCC, we selected genes that were methylated in at least two tumors in each group. Among the HBV-positive HCCs, 443 probe sets (corresponding to 332 unique genes) satisfied this criterion. Among the HCV-positive HCCs, 476 probe sets (342 unique genes) satisfied the criterion, and among the HBV/HCV-negative HCCs, 348 probe sets (259 unique genes) satisfied the criterion. Collectively, 714 probes (514 unique genes) were selected as commonly methylated genes. Of those, 137, 146, and 47 probe sets were methylated in only HBV-positive, HCV-positive, or HBV/HCV-negative HCC tissues, respectively (Fig. 1b). By contrast, a large number of genes were methylated in multiple categories, and 169 probe sets were methylated in all three groups (Fig. 1b). Consistent with the above results, unsupervised hierarchical clustering analysis demonstrated that some genes were methylated irrespective of the hepatitis virus status, and that HCV-positive HCCs exhibited the largest number of methylated genes (Fig. 1c, Supplementary Fig. 1). Gene ontology analysis of the commonly methylated genes revealed that genes related to “multicellular organismal process,” “developmental process,” and “system development” are significantly enriched among the methylated genes (Supplementary Table 2). In addition, pathway analysis suggested that some of the methylated genes are involved in differentiation and development (Supplementary Fig. 2).
Identification of novel genes methylated in HCC
Our MCAM analysis suggested that some genes were methylated in a hepatitis virus-specific manner, but a larger number were commonly methylated in HCC. Because recent studies have suggested that aberrant DNA methylation could be a useful diagnostic marker for HCC, we next aimed to identify novel genes frequently methylated in HCC. Among the genes commonly methylated irrespective of hepatitis virus status, we selected 14 (KLHL35, PAX5, PENK, SPDYA, LTBP2, DLX1, PGBD1, WNT9A, ADRA1A, RHOBTB1, GDNF, WNT11, MLL, and PLEC1) and carried out MSP to assess their methylation status in a series of HCC cell lines (Supplementary Fig. 3). We found that four (KLHL35, PAX5, PENK, and SPDYA) of the genes were frequently methylated in HCC cell lines, but showed only little or no methylation in normal liver tissue from a healthy individual (Supplementary Fig. 3). We therefore used quantitative bisulfite pyrosequencing to further analyze the methylation levels of these four genes (Supplementary Figs. 4 and 5).
To determine the extent to which these genes are aberrantly methylated in primary tumors, we analyzed a set of primary HCC specimens (HBV-positive, n = 21; HCV-positive, n = 21; HBV/HCV-negative, n = 15) and corresponding noncancerous liver tissues from the same patients (HBV-positive, n = 18; HCV-positive, n = 18; HBV/HCV-negative, n = 14). Bisulfite pyrosequencing analysis revealed the methylation levels of the four genes to be significantly higher in tumor tissues than in their noncancerous counterparts (KLHL35, 37.9 vs. 10.4 %, P < 0.001; PAX5, 23.4 vs. 7.7 %, P < 0.001; PENK 41.1 vs. 22.0 %, P < 0.001; SPDYA, 49.7 vs. 19.3 %, P < 0.001) (Supplementary Fig. 6). Moreover, these genes were frequently methylated in HCCs, irrespective of the hepatitis virus infection (KLHL35, HBV-positive, 37.5 vs. 9.9 %, P < 0.001; HCV-positive, 38.3 vs. 9.0 %, P < 0.001; HBV/HCV-negative, 37.7 vs. 13.0 %, P < 0.001; PAX5, HBV-positive, 22.2 vs. 7.6 %, P = 0.014; HCV-positive, 26.5 vs. 7.2 %, P < 0.001; HBV/HCV-negative, 20.7 vs. 8.5 %, P = 0.017; PENK, HBV-positive, 45.1 vs. 20.9 %, P < 0.001; HCV-positive, 39.2 vs. 23.5 %, P = 0.001; HBV/HCV-negative, 38.2 vs. 21.3 %, P = 0.006; SPDYA, HBV-positive, 51.5 vs. 19.8 %, P < 0.001; HCV-positive, 46.6 vs. 15.3 %, P < 0.001; HBV/HCV-negative, 51.3 vs. 23.7 %, P < 0.001) (Fig. 2a). The association between the methylation of each gene and the clinicopathological features are shown in Table 1. Methylation of KLHL35 and PAX5 was correlated with greater age, and SPDYA methylation was moderately correlated with higher PIVKA-II levels, but we found no other significant correlations (Table 1). We also generated an ROC curve and observed that methylation of the four genes discriminated strongly between tumor tissues and noncancerous liver tissue, suggesting that methylation of these genes could be a useful tumor marker (Fig. 2b). The most discriminating cutoffs for KLHL35, PAX5, PENK, and SPDYA were 14.8 % (sensitivity, 82.5 %; specificity, 88.0 %), 12.5 % (sensitivity, 63.2 %; specificity, 94.0 %), 28.4 % (sensitivity, 70.2 %; specificity, 96.0 %), and 30.3 % (sensitivity, 86.0 %; specificity, 94.0 %), respectively.

Quantitative methylation analysis of the genes identified by MCAM. a Summary of the bisulfite pyrosequencing analysis of KLHL35, PAX5, PENK, and SPDYA in tumor tissue (T) and noncancerous liver tissue (N) from HBV-positive, HCV-positive, and HBV/HCB-negative (NBNC) HCC patients. b ROC curve analysis of the methylation of the indicated genes. The area under the ROC curve (AUC) for each site conveys its utility (in terms of sensitivity and specificity) for distinguishing between HCC tissue and corresponding noncancerous liver tissue from the same HCC patients
Analysis of KLHL35, PAX5, PENK, and SPYDA methylation and expression
We next tested whether methylation of KLHL35, PAX5, PENK, and SPYDA was associated with their silencing in HCC. Bisulfite pyrosequencing analysis revealed that the degree to which these genes were methylated varied among the HCC cell lines, but it was always much higher than in normal liver tissue from a healthy individual (Fig. 3a). Quantitative RT-PCR analysis confirmed an inverse relationship between methylation and expression of KLHL35 and PAX5 in the cell lines and normal liver tissue (Fig. 3b), whereas methylation of PENK and SPDYA did not correlate significantly with their expression levels. The expression of PENK was undetectable in seven HCC cell lines and in normal liver tissue, irrespective of the methylation status (Fig. 3b). Conversely, although SPDYA was highly methylated in a majority of HCC cell lines, its expression was detectable in all cells, and most of the HCC lines exhibited greater SPDYA expression than did normal liver tissue (Fig. 3b). The above results suggest that KLHL35 and PAX5 are epigenetically silenced in HCC cells. Consistent with that idea, treating methylated cell lines with a DNA methyltransferase inhibitor, 5-aza-dC, restored the expression of KLHL35 and PAX5 (Fig. 3c). On the other hand, the expression of PENK and SPDYA does not appear to be affected by methylation.

Analysis of the methylation and expression of the indicated genes in HCC cell lines. a Bisulfite pyrosequencing of KLHL35, PAX5, PENK, and SPDYA in HCC cell lines and normal liver tissue from a healthy individual. b Quantitative RT-PCR of the four genes in HCC cell lines and normal liver tissue. c Quantitative RT-PCR of KLHL35 and PAX5 in HCC cell lines, with and without 5-aza-dC (aza) treatment
Analysis of LINE-1 methylation and its association with gene hypermethylation
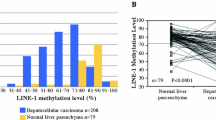
It was previously reported that LINE-1 is frequently hypomethylated in HCC, though most of those studies focused on HBV-positive tumors. Similarly, by using the bisulfite pyrosequencing, we found that levels of LINE-1 methylation were significantly lower in tumor tissues than in their noncancerous counterparts (48.5 vs. 66.8 %, P < 0.001). LINE-1 hypomethylation was prevalent, regardless of the tumor's hepatitis virus status, but the average methylation level was lowest in the HBV/HCV-negative tumors (HBV-positive, 50.8 vs. 66.3 %, P < 0.001; HCV-positive, 48.9 vs. 67.4 %, P < 0.001; HBV/HCV-negative, 44.7 vs. 66.6 %, P < 0.001; Fig. 4a). The ROC curve analysis revealed that LINE-1 methylation discriminated strongly between HCC tissue and noncancerous liver tissue (Fig. 4b), though no significant correlation was found between the levels of LINE-1 methylation and the clinicopathological characteristics of the samples (Table 1). Finally, we tested whether LINE-1 hypomethylation is linked to gene hypermethylation. We found an inverse relationship between the level of LINE-1 methylation and levels of KLHL35 and SPDYA methylation. On the other hand, we found no significant correlation between the LINE-1 hypomethylation and PAX5 or PENK methylation (Fig. 4c).

Analysis of LINE-1 methylation and its association with CpG island hypermethylation in HCC. a Summary of bisulfite pyrosequencing analysis of LINE-1 in tumor tissue (T) and corresponding noncancerous liver tissue (N) from HBV-positive, HCV-positive, and HBV/HCB-negative (NBNC) HCC patients. b ROC curve analysis of the utility of LINE-1 methylation for distinguishing between HCC tissue and corresponding noncancerous liver tissue from the same HCC patients. c Correlation between the level of LINE-1 methylation and methylation of the indicated genes in HCC tissues. The Pearson correlation coefficients and P values are shown
Discussion
In the present study, we carried out high-throughput CpG island methylation profiling in a set of primary HCC tissues with and without hepatitis virus infection. MCAM analysis enabled us to evaluate the methylation status of more than 6,000 gene promoters with high specificity and sensitivity [13]. Consistent with earlier studies that showed methylation to be more abundant in the HCV-positive HCCs than in the HBV-positive or hepatitis virus-negative HCCs [15, 18], we observed the highest number of methylated genes in HCV-positive HCC tissue. However, we also noted that a large number of genes were commonly methylated among HCCs, irrespective of the hepatitis virus status, indicating that aberrant methylation of multiple genes may be involved in a common mechanism underlying hepatocarcinogenesis. Moreover, studies have also shown that aberrant methylation detected in tissues or blood samples could be a useful biomarker for early detection of HCC [19, 20]. We therefore validated the methylation status of 14 genes and identified four genes that were frequently methylated in HCC tissues but showed little or no methylation in surrounding noncancerous tissues. The high-tumor specificity suggests that methylation of these genes may not occur at precancerous stages, such as chronic hepatitis or liver cirrhosis; instead, they may be acquired during malignant transformation.
The paired box 5 (PAX5) gene is a member of the paired box-containing family of transcription factors, which are involved in the control of organ development and tissue differentiation [21]. PAX5 is also known to be a B cell-specific activator protein that plays an essential role during B cell differentiation, neural development, and spermatogenesis. Methylation of the CpG island of PAX5 was first discovered in breast cancer cells using the MCA technique [22]. Subsequently, methylation and downregulation of PAX5 were found in lymphoid neoplasms [23]. In addition, while we are preparing the present manuscript, methylation of PAX5 was reported in HCC and gastric cancer [24, 25]. Restoration of PAX5 expression in HCC cells induced growth arrest and apoptosis through upregulation of various target genes, including p53, p21, and Fas ligand, suggesting that the PAX5 acts as a tumor suppressor [24].
The involvement of the kelch-like 35 (KLHL35) gene in cancer had not been reported until recently, when a genome-wide analysis of DNA methylation in renal cell carcinoma identified frequent hypermethylation of nine genes, including KHLH35 [26]. Although the function of the gene product remains unknown, RNAi-induced knockdown of KHLH35 in HEK293 cells promoted anchorage-independent growth, indicating its possible role in tumorigenesis [26].
The proenkephalin (PENK) gene encodes preproenkephalin, a precursor protein that is proteolytically cleaved to produce the endogenous opioid peptides met- and leu-enkephalin. Methylation of the CpG island of PENK was first identified in pancreatic cancer cells using the MCA technique [27]. Downregulated expression of PENK has also been reported in prostate cancer, suggesting its possible involvement in cancer development [28], and PENK methylation was recently identified in lung cancer, bladder cancer, and meningioma [29–31]. Although its functional role in cancer is not fully understood, a recent study showed that in response to cellular stress, PENK physically associates with p53 and RelA (p65) and regulates stress-induced apoptosis [32].
The SPDYA encodes Spy1, also known as Speedy, an atypical CDK activator known to promote cell survival, prevent apoptosis, and inhibit checkpoint activation in response to DNA damage [33]. The expression of SPDYA is upregulated in breast cancer [34], and its overexpression in a mouse model has been shown to accelerate mammary tumorigenesis [35]. Moreover, a recent study showed overexpression of SYPDA in HCC and its association with poor prognosis [36]. These results strongly suggest its involvement in oncogenesis. In the present study, we also observed that most of the HCC cell lines tested exhibited greater expression of SPDYA than normal liver tissue, regardless of the methylation status. Among the three transcription variants of SPDYA annotated in the NCBI Reference Sequence database, transcription start sites of variants 1 and 3 are located within the CpG island, while that of variant 2 are located approximately 5 kb downstream of the CpG island. Thus, the SPDYA transcript in HCC cells may be derived from the downstream transcription start site.
By analyzing the LINE-1 methylation levels, we and others have shown that global hypomethylation is a commonly observed feature of HCC [8, 9, 37]. Earlier studies have suggested that the association between global methylation and hepatitis status may be attributable to hepatitis B virus X protein, which can induce aberrant methylation of specific genes and global hypomethylation [38]. By contrast, we found in the present study that LINE-1 hypomethylation is prevalent among HCC tissues, regardless of the hepatitis virus infection, which suggests that global hypomethylation is involved in a common mechanism underlying hepatocarcinogenesis. It has been shown that the timing of global hypomethylation differs among tumor types. For example, hypomethylation is often observed during the early stages of colorectal and gastric carcinogenesis. By contrast, LINE-1 hypomethylation appears to be tumor-specific in HCC; it is rarely found in precancerous lesions such as chronic hepatitis or liver cirrhosis [8, 9]. A recent study showed that global hypomethylation is associated with a poorer prognosis in HCC patients [39]. In addition, the levels of serum LINE-1 hypomethylation in HCC patients reportedly correlate with serum HBs antigen status, large tumor size, and advanced tumor stage [40]. This suggests that hypomethylation may not occur at precancerous stages, and that LINE-1 methylation could be a useful biomarker with which to identify HCC and predict its clinical outcome.
The relationship between LINE-1 hypomethylation and CpG island hypermethylation in cancer is controversial. In one study, LINE-1 methylation levels were reduced in HCCs with the CpG island methylator phenotype, indicating a positive correlation between global hypomethylation and CpG island hypermethylation [9]. Another study showed that LINE-1 hypomethylation was positively correlated with hypermethylation of only a few genes (p16, CACNA1G, and CDKN1C), while methylation of a large number of genes showed inverse or no correlation with LINE-1 hypomethylation [12]. In the present study, we found that methylation of KLHL35 and SPYDA correlates positively with LINE-1 hypomethylation, whereas levels of PAX5 or PENK methylation are independent of LINE-1 methylation. These results suggest that the association between CpG island methylation and global hypomethylation may be site specific, and that hypomethylation of LINE-1 is a more generalized phenomenon than hypermethylation of CpG islands in HCC.
In summary, by screening targets of DNA methylation in HCC, we identified four frequently methylated genes. These genes are methylated in a cancer-specific manner and could be useful molecular markers for diagnosing HCC. In addition, we observed prevalent LINE-1 hypomethylation in HCC, irrespective of hepatitis virus infection. Identification of aberrant methylation in HCC may provide valuable information that not only contributes to our understanding of the pathogenesis of the disease, but also to the development of new strategies for diagnosis and therapy.
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
Chen CJ, Yu MW, Liaw YF. Epidemiological characteristics and risk factors of hepatocellular carcinoma. J Gastroenterol Hepatol. 1997;12:S294–308.
Montesano R, Hainaut P, Wild CP. Hepatocellular carcinoma: from gene to public health. J Natl Cancer Inst. 1997;89:1844–51.
Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46.
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703.
Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38.
Takai D, Yagi Y, Habib N, Sugimura T, Ushijima T. Hypomethylation of line1 retrotransposon in human hepatocellular carcinomas, but not in surrounding liver cirrhosis. Jpn J Clin Oncol. 2000;30:306–9.
Kim MJ, White-Cross JA, Shen L, Issa JP, Rashid A. Hypomethylation of long interspersed nuclear element-1 in hepatocellular carcinomas. Mod Pathol. 2009;22:442–9.
Kaneto H, Sasaki S, Yamamoto H, Itoh F, Toyota M, Suzuki H, Ozeki I, Iwata N, Ohmura T, Satoh T, Karino Y, Toyota J, Satoh M, Endo T, Omata M, Imai K. Detection of hypermethylation of the p16(ink4a) gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut. 2001;48:372–7.
Takagi H, Sasaki S, Suzuki H, Toyota M, Maruyama R, Nojima M, Yamamoto H, Omata M, Tokino T, Imai K, Shinomura Y. Frequent epigenetic inactivation of sfrp genes in hepatocellular carcinoma. J Gastroenterol. 2008;43:378–89.
Lee HS, Kim BH, Cho NY, Yoo EJ, Choi M, Shin SH, Jang JJ, Suh KS, Kim YS, Kang GH. Prognostic implications of and relationship between CpG island hypermethylation and repetitive DNA hypomethylation in hepatocellular carcinoma. Clin Cancer Res. 2009;15:812–20.
Gao W, Kondo Y, Shen L, Shimizu Y, Sano T, Yamao K, Natsume A, Goto Y, Ito M, Murakami H, Osada H, Zhang J, Issa JP, Sekido Y. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis. 2008;29:1901–10.
Arai E, Ushijima S, Gotoh M, Ojima H, Kosuge T, Hosoda F, Shibata T, Kondo T, Yokoi S, Imoto I, Inazawa J, Hirohashi S, Kanai Y. Genome-wide DNA methylation profiles in liver tissue at the precancerous stage and in hepatocellular carcinoma. Int J Cancer. 2009;125:2854–62.
Deng YB, Nagae G, Midorikawa Y, Yagi K, Tsutsumi S, Yamamoto S, Hasegawa K, Kokudo N, Aburatani H, Kaneda A. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 2010;101:1501–10.
Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, Gao W, An B, Fujii M, Murakami H, Osada H, Taniguchi T, Usami N, Kondo M, Hasegawa Y, Shimokata K, Matsuo K, Hida T, Fujimoto N, Kishimoto T, Issa JP, Sekido Y. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res. 2009;69:9073–82.
Suzuki H, Yamamoto E, Nojima M, Kai M, Yamano HO, Yoshikawa K, Kimura T, Kudo T, Harada E, Sugai T, Takamaru H, Niinuma T, Maruyama R, Yamamoto H, Tokino T, Imai K, Toyota M, Shinomura Y. Methylation-associated silencing of microrna-34b/c in gastric cancer and its involvement in an epigenetic field defect. Carcinogenesis. 2010;31:2066–73.
Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–18.
Wong IH, Zhang J, Lai PB, Lau WY, Lo YM. Quantitative analysis of tumor-derived methylated p16ink4a sequences in plasma, serum, and blood cells of hepatocellular carcinoma patients. Clin Cancer Res. 2003;9:1047–52.
Zhang YJ, Wu HC, Shen J, Ahsan H, Tsai WY, Yang HI, Wang LY, Chen SY, Chen CJ, Santella RM. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res. 2007;13:2378–84.
Carotta S, Holmes ML, Pridans C, Nutt SL. Pax5 maintains cellular identity by repressing gene expression throughout B cell differentiation. Cell Cycle. 2006;5:2452–6.
Palmisano WA, Crume KP, Grimes MJ, Winters SA, Toyota M, Esteller M, Joste N, Baylin SB, Belinsky SA. Aberrant promoter methylation of the transcription factor genes pax5 alpha and beta in human cancers. Cancer Res. 2003;63:4620–5.
Lazzi S, Bellan C, Onnis A, De Falco G, Sayed S, Kostopoulos I, Onorati M, D’Amuri A, Santopietro R, Vindigni C, Fabbri A, Righi S, Pileri S, Tosi P, Leoncini L. Rare lymphoid neoplasms coexpressing B- and T-cell antigens. The role of pax-5 gene methylation in their pathogenesis. Hum Pathol. 2009;40:1252–61.
Liu W, Li X, Chu ES, Go MY, Xu L, Zhao G, Li L, Dai N, Si J, Tao Q, Sung JJ, Yu J. Paired box gene 5 is a novel tumor suppressor in hepatocellular carcinoma through interaction with p53 signaling pathway. Hepatology. 2011;53:843–53.
Li X, Cheung KF, Ma X, Tian L, Zhao J, Go MY, Shen B, Cheng AS, Ying J, Tao Q, Sung JJ, Kung HF, Yu J. Epigenetic inactivation of paired box gene 5, a novel tumor suppressor gene, through direct upregulation of p53 is associated with prognosis in gastric cancer patients. Oncogene. 2011. doi:10.1038/onc.2011.511.
Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H, Brown M, Kishida T, Yao M, Banks RE, Clarke N, Latif F, Maher ER. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30:1390–401.
Ueki T, Toyota M, Skinner H, Walter KM, Yeo CJ, Issa JP, Hruban RH, Goggins M. Identification and characterization of differentially methylated cpg islands in pancreatic carcinoma. Cancer Res. 2001;61:8540–6.
Goo YA, Goodlett DR, Pascal LE, Worthington KD, Vessella RL, True LD, Liu AY. Stromal mesenchyme cell genes of the human prostate and bladder. BMC Urol. 2005;5:17.
Chung JH, Lee HJ, Kim BH, Cho NY, Kang GH. DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Arch. 2011;459:201–11.
Chung W, Bondaruk J, Jelinek J, Lotan Y, Liang S, Czerniak B, Issa JP. Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments. Cancer Epidemiol Biomarkers Prev. 2011;20:1483–91.
Kishida Y, Natsume A, Kondo Y, Takeuchi I, An B, Okamoto Y, Shinjo K, Saito K, Ando H, Ohka F, Sekido Y, Wakabayashi T. Epigenetic subclassification of meningiomas based on genome-wide DNA methylation analyses. Carcinogenesis. 2012;33:436–41.
McTavish N, Copeland LA, Saville MK, Perkins ND, Spruce BA. Proenkephalin assists stress-activated apoptosis through transcriptional repression of nf-kappab- and p53-regulated gene targets. Cell Death Differ. 2007;14:1700–10.
Gastwirt RF, McAndrew CW, Donoghue DJ. Speedy/ringo regulation of CDKs in cell cycle, checkpoint activation and apoptosis. Cell Cycle. 2007;6:1188–93.
Zucchi I, Mento E, Kuznetsov VA, Scotti M, Valsecchi V, Simionati B, Vicinanza E, Valle G, Pilotti S, Reinbold R, Vezzoni P, Albertini A, Dulbecco R. Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc Natl Acad Sci U S A. 2004;101:18147–52.
Golipour A, Myers D, Seagroves T, Murphy D, Evan GI, Donoghue DJ, Moorehead RA, Porter LA. The spy1/ringo family represents a novel mechanism regulating mammary growth and tumorigenesis. Cancer Res. 2008;68:3591–600.
Ke Q, Ji J, Cheng C, Zhang Y, Lu M, Wang Y, Zhang L, Li P, Cui X, Chen L, He S, Shen A. Expression and prognostic role of spy1 as a novel cell cycle protein in hepatocellular carcinoma. Exp Mol Pathol. 2009;87:167–72.
Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61:4238–43.
Park IY, Sohn BH, Yu E, Suh DJ, Chung YH, Lee JH, Surzycki SJ, Lee YI. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology. 2007;132:1476–94.
Calvisi DF, Simile MM, Ladu S, Pellegrino R, De Murtas V, Pinna F, Tomasi ML, Frau M, Virdis P, De Miglio MR, Muroni MR, Pascale RM, Feo F. Altered methionine metabolism and global DNA methylation in liver cancer: relationship with genomic instability and prognosis. Int J Cancer. 2007;121:2410–20.
Tangkijvanich P, Hourpai N, Rattanatanyong P, Wisedopas N, Mahachai V, Mutirangura A. Serum line-1 hypomethylation as a potential prognostic marker for hepatocellular carcinoma. Clin Chim Acta. 2007;379:127–33.
Acknowledgments
We thank Dr. Yutaka Kondo for technical advice on MCAM analysis and Masami Ashida for technical assistance. This study was supported in part by a Grant-in-Aid for Scientific Research (B) from the Japan Society for Promotion of Science (Y. Shinomura), a Grant-in-Aid for the Third-term Comprehensive 10-year Strategy for Cancer Control (M. Toyota and H. Suzuki), and a Grant-in-Aid for Cancer Research from the Ministry of Health, Labor, and Welfare, Japan (M. Toyota and H. Suzuki).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Primer sequences used in this study (XLS 36 kb)
Supplementary Table 2
GO analysis of commonly methylated genes in HCC (XLS 29 kb)
ESM 1
(PDF 936 kb)
Rights and permissions
About this article
Cite this article
Shitani, M., Sasaki, S., Akutsu, N. et al. Genome-wide analysis of DNA methylation identifies novel cancer-related genes in hepatocellular carcinoma. Tumor Biol. 33, 1307–1317 (2012). https://doi.org/10.1007/s13277-012-0378-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-012-0378-3