
Abstract
Changes in DNA methylation patterns are an important characteristic of human cancer including lung cancer. In particular, hypermethylation of CpG islands is a signature of malignant progression. Methylated CpG islands are promising diagnostic markers for the early detection of cancer. However, the full extent and sequence context of DNA hypermethylation in lung cancer has remained unknown. We have used the methylated CpG island recovery assay and high-resolution microarray analysis to find hypermethylated CpG islands in squamous cell carcinomas (SCC) and adenocarcinomas of the lung. Each tumor contained several hundred hypermethylated CpG islands. In an initial microarray screen, 36 CpG islands were methylated in five of five (=100%) of the SCC tumors tested and 52 CpG islands were methylated in at least 75% of the adenocarcinomas tested (n = 8). Using sodium-bisulfite-based approaches, 12 CpG islands (associated with the BARHL2, EVX2, IRX2, MEIS1, MSX1, NR2E1, OC2, OSR1, OTX1, PAX6, TFAP2A, and ZNF577 genes) were confirmed to be methylated in 85% to 100% of the squamous cell carcinomas and 11 CpG islands (associated with the CHAD, DLX4, GRIK2, KCNG3, NR2E1, OSR1, OTX1, OTX2, PROX1, RUNX1, and VAX1 genes) were methylated in >80% of the adenocarcinomas. From the list of genes that were methylated in lung adenocarcinomas, we identified the gene FAT4 and found that this gene was methylated in 39% of the tumors. FAT4 is the closest mammalian homologue of the Drosophila tumor suppressor Fat which is an important component of the Hippo growth control pathway. Many of these newly discovered methylated CpG islands hold promise for becoming biomarkers for the early detection of lung cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
5-Methylcytosine, present at 70–80% of all CpG dinucleotides, is the major modified base found in mammalian DNA. It has been known for some time that the level of 5-methylcytosine is globally reduced in tumor tissues relative to corresponding normal tissues [14, 16, 36], mostly due to hypomethylation of different classes of repetitive DNA sequences [34]. It was also observed that gene-specific hypermethylation at CpG-rich, so-called CpG-island sequences occurs specifically in cancer tissues [2]. CpG island hypermethylation is a phenomenon commonly observed during the development and progression of human tumors [21]. In the 1990s, researchers reported hypermethylation of CpG islands of several known tumor suppressor genes and other genes involved in important growth control or genome defense pathways, for example DNA repair genes [8, 12, 17, 20, 21, 23, 24, 27]. Today, there are many reports documenting methylation of CpG islands associated with a large number of different genes, including almost every type of human malignancy. In lung cancer, many specific CpG islands are methylated, for example those associated with the genes CDKN2A, RASSF1A, RARbeta, MGMT, GSTP1, CDH13, APC, DAPK, TIMP3, and a number of others [1, 9, 11, 42, 47, 49]. The methylation frequency of a particular CpG island (defined as the percentage of tumors analyzed in a study that carry methylated alleles) generally ranges from less than 10% to over 80% of the tumors depending on the histological subtype of tumor, the study population, and/or the methodology used to assess and quantitate DNA methylation.
Detection of methylated CpG islands in accessible biological materials such as serum or sputum has the recognized potential to be useful for the early detection and diagnosis of cancer, including lung cancer [3, 25, 43]. Highly sensitive techniques that can clearly detect the methylated tumor-associated DNA fragments among a large excess of unmethylated molecules are necessary to accomplish this goal. Most useful as DNA methylation markers would only be those genes that show close to background levels of methylation in normal human tissues and those that are methylated in a large fraction of the tumors.
Current research approaches in cancer epigenetics are focused on the characterization of the complete set of DNA methylation changes in cancer. Several techniques have been introduced and have been summarized comprehensively [13]. We recently developed a useful method, the methylated CpG island recovery assay (MIRA) that—unlike most other approaches—does not depend on the use of sodium bisulfite, restriction enzymes, or antibodies for identifying the methylated regions [31]. The MIRA method is based on the high affinity of the MBD2b/MBD3L1 protein complex for methylated CpG dinucleotides. For efficient pull-down of methylated DNA by this method, two or more methylated CpG sites in a DNA fragment of 50 base pairs or less are required [32]. MIRA is compatible with microarray analysis [32] or high-throughput DNA sequencing [35]. We have used the MIRA method in combination with high-resolution CpG island microarrays to characterize the full extent of DNA methylation changes that occur at CpG islands in non-small cell lung cancer tissues.
DNA methylation analysis of lung cancer
To analyze tumor-associated DNA methylation changes, we compared stage I lung squamous cell carcinomas (SCCs) or adenocarcinomas (AC) to normal matched lung tissues [34]. We used the MIRA-assisted microarray method for DNA methylation analysis [32, 33]. MIRA-enriched methylated DNA fractions obtained from tumor tissue and from matching normal lung tissue removed with surgery were analyzed on Agilent CpG island arrays covering a total of 27,800 CpG islands.
A complete set of hypermethylated CpG islands and discovery of new DNA methylation biomarkers
Five stage I squamous cell carcinomas and eight stage I adenocarcinomas of the lung were analyzed on these arrays along with matched normal lung. Using the criteria and cutoffs defined previously [34], the number of methylated CpG islands ranged from 216 to 744 in the five individual squamous cell tumors (Table 1). For adenocarcinomas, between 219 and 908 CpG islands were methylated per tumor (Table 1).
Squamous cell carcinomas
Using MIRA-assisted microarray analysis, we identified 36 CpG islands that were methylated in all of the five SCC tumors analyzed (Fig. 1 and Table 2). A large fraction of the methylated CpG islands were mapped to homeobox genes in the genome. Since these 36 loci had excellent potential to be specific and sensitive methylation biomarkers for SCC, we analyzed 12 randomly chosen markers (BARHL2, EVX2, IRX2, MEIS1, MSX1, NR2E1, OC2, OSR1, OTX1, PAX6, TFAP2A, and ZNF577) in a larger series of 20 SCCs [34] by bisulfite-based COBRA assays [46]. This assay is semi-quantitative, technically robust and is commonly used for assessing the methylation status of CpG islands in smaller to medium size sample series; this method has a very low rate of false positive results. The methylation frequency of the individual markers ranged from 17/20 (=85%) to 20/20 (=100%) of the tumors. The OTX1 and NR2E1 associated CpG islands were methylated in all SCC tumors tested (=100%). Several of these SCC markers were highly specific for tumor-associated methylation, i.e., little or no detectable methylation was observed in tumor-adjacent normal lung tissue or in the lungs of non-tumor patients. These included the CpG islands of the OTX1, BARHL2, MEIS1, OC2, TFAP2A, and EVX2 genes [34]. None of these CpG islands was methylated substantially in white blood cell DNA from healthy individuals.

Methylation of CpG islands in lung squamous cell carcinomas. The red bars indicate methylation of individual CpG islands across a series of five stage I lung squamous cell carcinomas. The CpG islands methylated in all five tumors are marked by arrows
Adenocarcinomas
Using MIRA-assisted microarray analysis, we identified 52 CpG islands that were methylated in at least six out of eight adenocarcinomas (Table 3). Several of these adenocarcinoma methylation markers (CHAD, DLX4, GRIK2, KCNG3, NR2E1, OSR1, OTX1, OTX2, PROX1, RUNX1, and VAX1) were chosen at random for verification by bisulfite-based COBRA assays. These selected adenocarcinoma markers were methylated in more than 80% of the tumors (Fig. 2). The CHAD gene was methylated in 8 of 11 tumors tested (data not shown). None of these CpG islands was substantially methylated in blood DNA from healthy individuals or in non-cancerous lung DNA. The tumor specificity and high methylation frequency of these genes makes them excellent candidates for use in future diagnostic applications for early detection of lung cancer.

Verification of DNA methylation markers in normal lung tissue and in matching adenocarcinoma samples. Methylation differences between adenocarcinomas (T) and matching normal pairs (N) were detected by COBRA assays for the indicated gene targets. “–” refers to control digestion with no BstUI, “+” refers to BstUI-digested samples. Digestion by BstUI indicates methylation of the sequence tested (5′CGCG). The indicated CpG islands were analyzed (see Table 3 for chromosomal location of the CpG islands)
Methylation of FAT4 encoding a component of the Hippo tumor suppressor pathway in lung adenocarcinomas
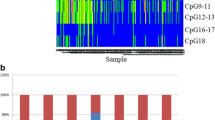
One of the methylated targets in adenocarcinomas and SCCs was the gene FAT4. To confirm the microarray results, we analyzed methylation of the FAT4-associated promoter CpG island in 18 stage I adenocarcinomas and matched normal lung tissue (Fig. 3a). FAT4 was methylated in 7/18 tumors (=39%). We also confirmed that FAT4 is silenced in a series of non-small cell lung tumors compared to corresponding normal tissue when a cDNA panel from Origene, which contains various types of non-small cell lung tumors of different stages (stage I and II), was used. According to this RT-PCR analysis, expression of FAT4 was reduced in 18 of 23 stages I and II lung tumors relative to matched normal lung tissue (Fig. 3b) suggesting that methylation of this gene is perhaps only one mechanism leading to reduced expression in tumors. FAT4 encodes the closest mammalian homologue of the Drosophila Fat protocadherin (37% identical at the amino acid level). Fat is an upstream component of the Hippo pathway in flies [6]. The Hippo pathway is a signaling cascade involved in organ size control, tumor suppression, and apoptosis [29, 39]. Mutations of this pathway in mice has been linked to tumorigenesis [26, 40, 48]. However, it has been difficult to clearly demonstrate functional inactivation of this tumor suppressor pathway in human tumors. Mutations in Hippo pathway components, such as the MST and LATS kinases, are very infrequent. Epigenetic inactivation is seen most commonly for RASSF1A, a regulator of MST kinases [18, 19, 30] and sometimes for MST genes themselves [38]. In lung tumors, RASSF1A is most frequently inactivated by promoter methylation in small cell lung cancers [10]. In non-small cell lung cancers, RASSF1A methylation occurs at a frequency of 30–40% in adenocarcinomas and squamous cell carcinomas [5, 9]. We have not yet tested if methylation of RASSF1A and FAT4, both being upstream regulators of the Hippo pathway, are mutually exclusive or cooperate in lung tumors. Interestingly, Fat4 has recently been identified as a susceptibility gene for pulmonary adenomas in mice [4]. Thus, methylation of the promoter of FAT4 may have functional consequences for tumor initiation or progression in the lung. In this sense, methylation of FAT4 can be considered a potential epigenetic “driver” event for tumorigenesis rather than just a “passenger” event that would be without functional consequences [22].

Methylation of the FAT4 gene in lung adenocarcinomas. a Methylation of the FAT4 promoter-associated CpG island was tested in 18 stage I lung adenocarcinomas (T) and matched normal lung tissue (N). Methylation differences were detected by COBRA assays. “–” refers to control digestion with no BstUI, “+” refers to BstUI-digested samples. Digestion by BstUI indicates methylation of the sequence tested (5′CGCG), which was found in seven of the tumors. b FAT4 silencing in a series of non-small cell lung tumors. Normalized cDNA from 23 paired lung tumor/normal samples were tested with TaqMan qRT-PCR and the expression level of FAT4 was derived from comparing with β-actin internal control with two highly expressed samples set as 1
DNA methylation markers for lung cancer
The novel aspect of our work has been the comprehensive methylation analysis of all CpG islands in human lung cancer using microarrays [32–34]. We were able to directly measure the methylation levels at over 27,000 CpG islands and found that between approximately 200 and 900 of these islands were methylated in individual lung SCC and AC samples (Table 1). These numbers are compatible with earlier estimates derived from analysis of only a subset of CpG islands methylated in cancer [7]. It is clear, of course, that not all of these genes can be tumor suppressor genes or in other ways can be driver events for tumorigenesis. For example, our earlier observations indicated that a substantial subset of the methylated genes (20–40% depending on the individual tumor) were homeobox genes [32, 33]. Homeobox genes are regulated by the Polycomb complex, and it is a common observation that Polycomb-associated genes are frequently methylated in cancer [28, 33, 37, 41, 44, 45]. Homeobox gene-associated CpG islands were among the most useful DNA methylation markers identified in lung cancer. The CpG islands of the OTX1, BARHL2, MEIS1, OC2, TFAP2A, and EVX2 genes were tumor-specifically methylated in SCC with little or no detectable methylation seen in normal lung tissue or in blood DNA [34]. Importantly, the methylation frequency of these markers (85% to 100% of the tumors were methylated) is much higher than methylation frequencies of most other lung cancer DNA methylation markers reported previously. For example, OTX1 was tumor specifically methylated in 20/20 (=100%) of the SCC tumors. For adenocarcinomas, several promising markers have been identified including CHAD, DLX4, GRIK2, KCNG3, NR2E1, OSR1, OTX1, OTX2, PROX1, RUNX1, and VAX1. Methylation of these genes in lung cancer has not yet been reported, except for RUNX1 [15]. Very commonly, frequently methylated genes are methylated in both subtypes of non-small cell lung cancers. For example, the CpG islands associated with the NR2E1, OSR1, and OTX1 genes were methylated in both adenocarcinomas and squamous cell carcinomas at a frequency of over 95%. These markers are excellent candidates for future clinical or diagnostic applications aimed at either detection of early disease in body fluids such as blood or sputum, or at disease management and follow-up using molecular diagnostic testing.
References
Anglim PP, Alonzo TA, Laird-Offringa IA. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol Cancer. 2008;7:81.
Baylin SB, Hoppener JW, de Bustros A, Steenbergh PH, Lips CJ, Nelkin BD. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res. 1986;46:2917–22.
Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17.
Berndt A, Cario CL, Silva KA, Kennedy VE, Harrison DE, Paigen B, Sundberg JP. Identification of fat4 and tsc22d1 as novel candidate genes for spontaneous pulmonary adenomas. Cancer Res. 2011;71:5779–91.
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001;93:691–9.
Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. Delineation of a Fat tumor suppressor pathway. Nat Genet. 2006;38:1142–50.
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O’Dorisio MS, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8.
Costello JF, Plass C. Methylation matters. J Med Genet. 2001;38:285–303.
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–9.
Dammann R, Takahashi T, Pfeifer GP. The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene. 2001;20:3563–7.
Dammann R, Strunnikova M, Schagdarsurengin U, Rastetter M, Papritz M, Hattenhorst UE, Hofmann HS, Silber RE, Burdach S, Hansen G. CpG island methylation and expression of tumour-associated genes in lung carcinoma. Eur J Cancer. 2005;41:1223–36.
Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–9.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92.
Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, Dong ZM, Jordan CD, Kiviat NB, Vesselle H. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17:645–54.
Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94.
Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 1995;55:4531–5.
Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer GP. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr Biol. 2007;17:700–5.
Guo C, Zhang X, Pfeifer GP. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem. 2011;286:6253–61.
Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–30.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92.
Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet. 2010;70:277–308.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11.
Lahtz C, Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol. 2011;3:51–8.
Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66.
Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, Lim DS. The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:8248–53.
Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–92.
Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505.
Pfeifer GP, Dammann R, Tommasi S. RASSF proteins. Curr Biol. 2010;20:R344–5.
Rauch T, Pfeifer GP. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab Investig. 2005;85:1172–80.
Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–47.
Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci U S A. 2007;104:5527–32.
Rauch TA, Zhong X, Wu X, Wang M, Kernstine KH, Wang Z, Riggs AD, Pfeifer GP. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci U S A. 2008;105:252–7.
Rauch TA, Pfeifer GP. DNA methylation profiling using the methylated-CpG island recovery assay (MIRA). Methods. 2010;52:213–7.
Riggs AD, Jones PA. 5-Methylcytosine, gene regulation, and cancer. Adv Cancer Res. 1983;40:1–30.
Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6.
Seidel C, Schagdarsurengin U, Blumke K, Wurl P, Pfeifer GP, Hauptmann S, Taubert H, Dammann R. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog. 2007;46:865–71.
Sudol M, Harvey KF. Modularity in the Hippo signaling pathway. Trends Biochem Sci. 2010;35:627–33.
Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 2005;65:92–8.
Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11:R14.
Topaloglu O, Hoque MO, Tokumaru Y, Lee J, Ratovitski E, Sidransky D, Moon CS. Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Clin Cancer Res. 2004;10:2284–8.
Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer. 2005;5:223–31.
Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8.
Wu X, Rauch TA, Zhong X, Bennett WP, Latif F, Krex D, Pfeifer GP. CpG island hypermethylation in human astrocytomas. Cancer Res. 2010;70:2718–27.
Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–4.
Yanagawa N, Tamura G, Oizumi H, Takahashi N, Shimazaki Y, Motoyama T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003;94:589–92.
Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–38.
Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249–55.
Acknowledgments
This work was supported by NIH grant CA084469 and a grant from the University of California Tobacco Related Disease Research Program (TRDRP) (to G.P.P.)
Conflicts of interest
Under a licensing agreement between City of Hope and Active Motif (Carlsbad, CA), the methylated CpG island recovery assay (MIRA) technique was licensed to Active Motif, and the authors T.A.R. and G.P.P. are entitled to a share of the royalties received by City of Hope from sales of the licensed technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tibor A. Rauch and Zunde Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Rauch, T.A., Wang, Z., Wu, X. et al. DNA methylation biomarkers for lung cancer. Tumor Biol. 33, 287–296 (2012). https://doi.org/10.1007/s13277-011-0282-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-011-0282-2