
Abstract
Pancreatic cancer is one of the most malignant diseases in the world. Interferon regulator factor 2 (IRF-2), an interferon regulatory factor, has been known to act as an oncogene in distinct types of cancer. In this study, we found that the expression of IRF-2 was up-regulated in primary pancreatic cancer samples and associated with tumor size, differentiation, tumor–node–metastasis stage, and survival of the patients. In pancreatic cancer cells, knockdown on the expression of IRF-2 inhibited cell growth in the liquid culture and on the soft agar. Mechanistically, IRF-2 modulated the growth of pancreatic cancer cells through regulating proliferation and apoptosis effectors, such as cyclin D1 and BAX. Collectively, these results suggest that IRF-2 plays an important role in the tumorigenesis of pancreatic cancer and down-regulation of IRF-2 would be a new treatment target for pancreatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is one of the most aggressive malignant cancers ranking eighth as the cause of cancer-related death in the world [1, 2]. Although the technology of diagnose and therapy of pancreatic cancer in recent years was improved, survival rate and median survival of the patients are still pessimistic [3–6], which is mainly due to the difficulty in detecting at the early stage and the complicated pathological mechanism [5, 7–9]. Tumorigenesis of pancreatic cancer is attributed to dysregulation of multiple genes. For example, K-ras activation and lose of critical tumor suppressors including p53 and INK4a have been found to play important role in the malignant transformation of pancreatic tumor [9–12].
Interferon regulator factor 2 (IRF-2) belongs to the family of interferon regulatory factors, which has been known to be implicated in tumorigenesis through regulating the expression of target genes [13–15]. As a transcriptional factor, IRF-2 was regarded as the functional antagonist of IRF-1 and could repress or activate the transcription of its downstream target genes containing interferon regulatory factor element binding site in the promoter [14, 16, 17]. Recently, more and more evidence uncovered the pivotal role of IRF-2 in tumor progression [18, 19]. By suppressing the function of p21, IRF-2 promoted cell growth in leukemogenesis, and knockdown of IRF-2 blocked leukemia cell growth [20, 21]. Elevated expression of IRF-2 was found in breast cancer, which was positively correlated to the tumor stage [22–24]. Knockdown of IRF-2 in esophageal squamous cell carcinoma (ESCC) cells decreased the expression levels of cyclin D1 and activated caspase-9, while over-expression of IRF-2 in ESCC cells enhanced tumorigenicity, which was reversed by up-regulation of IRF-1 [25, 26]. Several studies have implicated the tumor promoting effects of IRF-2 in pancreatic cancer; however, the expression of IRF-2 in the clinical samples of pancreatic cancer and the mechanism for IRF-2 to promote pancreatic cancer is not clear [17, 27]. Here, in pancreatic cancer species, we found that the up-regulation of IRF-2 in primary pancreatic cancer samples and the expression of IRF-2 were associated with tumor size, differentiation, tumor–node–metastasis (TNM) stage and survival of pancreatic cancer patients. Silencing the expression of IRF-2 inhibited cell growth in the liquid culture and on the soft agar. Furthermore, we found that IRF-2 modulates cell growth of pancreatic cancer cells through regulating the proliferation and apoptosis effectors, such as cyclin D1 and BAX. Taken together, these results suggest the oncogenic role of IRF-2 in pancreatic cancer.
Materials and methods
Pancreatic cancer tissue samples
Thirty pairs of fresh pancreatic cancer samples and their corresponding normal tissues which were at least 3 cm away from the tumor were obtained from pancreatic ductal adenocarcinoma patients treated at Zhongshan Hospital of Fudan University (Shanghai, China) from 2009 to 2010, and none of the patients received any neoadjuvant therapy. All dissected samples were frozen immediately after surgery and stored at −80°C until needed. The mRNA level of IRF-2 in these paired tissues was analyzed using reverse transcriptase polymerase chain reaction (PCR). The protein level of IRF-2 in seven randomly selected paired tissues from the samples above was analyzed using western blot. Paraffin-embedded samples of pancreatic ductal adenocarcinoma from 156 patients, who underwent surgical excision and without any prior therapy between 2005 and 2010 at Zhongshan Hospital of Fudan University, were included in the study of tissue array. All patients had given informed consent. The study was approved by the Institutional Review Board of Zhongshan Hospital of Fudan University.
Cell culture
Pancreatic cancer cell line MIAPaCa-2 and PANC-1 was purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai Institute of Cell Biology, Chinese Academy of Sciences) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (Gibco), 10 U/ml penicillin-G, and 10 U/ml streptomycin. Cells were incubated at 37°C in 5% CO2 atmosphere.
Antibodies
Anti-IRF-2 antibody, anti-cyclin D1 antibody, anti-poly-ADP-ribose polymerase (anti-PARP) antibody, anti-BAX antibody, anti-caspase-8 antibody, anti-proliferating cell nuclear antigen (anti-PCNA) antibody, anti-tubulin antibody, and anti-glyceraldehyde 3-phosphate dehydrogenase antibody were purchased from Santa Cruz Biotechnology; horseradish peroxidase-labeled secondary antibodies were purchased from Cell Signaling Technology.
Tissue array
Tissue array was constructed using BIONAN Tissue arrayer TMA600 (BIONAN Biotechnology, Shanghai, China) according to the manufacturer’s protocols. Briefly, representative areas, tumor tissues and matching normal tissues, were premarked in the paraffin-embedded blocks by H&E staining. Duplicates of 3-mm-diameter cylinders from the center of the tumor as well as matching normal tissues were included in each case to ensure reproducibility and homogeneity. Sections 4 μm thick were placed on slides coated with 3-aminopropyltriethoxysilane.
Immunohistochemistry and scoring system
Immunohistochemistry (IHC) staining was performed according to the DAB two-step kit protocol (R&D Systems, cell & tissue staining kit). Briefly, slides were deparaffinized in xylene and hydrated through a graded alcohol series before being placed in 0.3% H2O2–phosphate-buffered saline (PBS) blocking solution to inhibit endogenous peroxidase activity. Then slides were incubated with IRF-2 antibody (1:300, Santa Cruz) at 4°C overnight and treated with secondary antibody for 40 min at room temperature. The sections were developed in diaminobenzidine solution under a microscope and counterstained with hematoxylin. Negative control slides omitting the primary antibody were included in all assays.
Arrays were evaluated at ×200 magnification light microscopy by two pathologists blinded to the clinicopathologic data of the patients. The intensity of staining was rated as either 0 (no signal as the negative controls), 1 (weak), or 2 (strong); the percentage of positive tumor cells was graded as 0 (no cells), 1 (1–25% of total tumor cells), 2 (26–50%), 3 (51–75%), and 4 (75–100%). The immunoreactive score for whole slides was calculated by multiplying the score of percentage positive cells and the score of staining intensity. According to comparison of IHC scores of IRF-2 in tumor with matched normal tissues, two levels about tumor tissues (low or high) were determined by the pathologists. The tumor tissues with scores ≥3 were referred as high, and those with scores <3 were low.
Real-time PCR
The mRNA level of IRF-2 and β-actin was analyzed by real-time PCR. Briefly, total RNA was isolated from tissues of pancreatic cancer patients using TRIzol reagent (Invitrogen), and their quality was then determined by visibility of 18S and 28S RNA bands under UV light. Two micrograms of total RNA with high quality was processed directly to cDNA with the reverse transcription kit (Promega, Madison, WI, USA), following the standard instructions, in a total volume of 25 μl. Amplification reactions were performed in a 20-μl volume of the LightCycler-DNA Master SYBR Green I mixture (Roche Applied Science). PCR reaction included components as follows: 10 pmol of primer, 2 mM MgCl2, 200 μM dNTP mixture, 0.5 U of Taq DNA polymerase, and universal buffer. The thermal cycling conditions were as follows: 95°C for 3 min; 40 cycles of 95°C for 30 s, 58°C for 20 s, and 72°C for 30 s; and 72°C for 10 min. The specificity of amplification was examined by melting curve analysis and electrophoresis in 2% agarose gel. All of the reactions were performed in triplicate in an iCycler iQSystem (Bio-Rad). The primer sequences for the human IRF-2 gene were as follows: forward primer, 5′-TGGATGCATGCGGCTAGA-3′; reverse primer: 5′-CATCTGAAATTCGCCTTCC-3′. As an internal standard, a fragment of human β-actin was amplified by PCR using the following primers: forward primer, 5′-GATCATTGCTCCTCCTGAGC-3′, and reverse primer, 5′-ACTCCTGCTTG CTGATCCAC-3′. Data were presented as the fold change of IRF-2 expression in each tumor tissue relative to its paired normal sample after normalization with β-actin.
Western blot analysis
Cells were harvested, washed twice with PBS, and lysed in RIPAII buffer (500 mM NaCl; 50 mM Tris, pH 7.4; 0.1% SDS; 1% NP-40; 0.5% Na-DOC; 0.05% NaN3; complete protease inhibitor mix) for 30 min on ice. Cells or tissue lysates were centrifuged at 12,000×g for 15 min, and the supernatants were collected. Protein were separated on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane (Millipore, Bedford, MA, USA), and incubated with antibodies. The immunoreactive protein bands were visualized by ECL kit (Pierce).
RNA interference
Target sequence for IRF-2 small interfering RNA was as listed: 1#5′-GTGGATAGTACGGTGAACA-3′ or 2#5′-GGACCAACAAGGGCAGTGG-3′. The control nucleotide sequence of small interfering RNA was 5′-GAATGCGAGCGAGCGAGCA-3′, which was the random sequence that was not related to IRF-2 mRNA. FG12 RNAi vector was used to produce small double-stranded RNA (small interfering RNA) to inhibit target gene expression in pancreatic cancer cells. FG12 vector with IRF-2 siRNA or IRF-2 siRNA con was transfected into 293T, and the virus with IRF-2 siRNA or IRF-2 siRNA con was harvested from culture medium. The harvested virus was purified by centrifugation at 25,000×g (4°C, 150 min), and appropriate amounts of virus were used to infect MIAPaCa-2 cells and PANC-1 cells. After 3 days of infection, the GFP-positive cells were sorted by flow cytometry (BD Biosciences), which all stably expressed IRF-2 siRNA or IRF-2 siRNA con.
MTT assay and BrdU labeling
In 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, cells were seeded into 96-well plates (2 × 103 cells/well) and grown for the various length of time. To measure cell growth, 20 μl 5 mg/ml MTT was added into the media and cultured at 37°C. After 5 h, 200 μl DMSO was added to resolve the generated formazan right after removing the cell medium, and the OD540 value of the solvent was measured by an automatic microplate reader. The measurement process was performed every 24 h for 7 or 8 days to generate a cell growth curve, and each experiment was repeated at least three times.
In bromodeoxyuridine (BrdU) labeling, BrdU pulse labeling was performed in MIAPaCa-2 cells stably transfected with plasmids expressing siRNA of IRF-2 and siRNA con. The BrdU incorporation was examined by using antibody against BrdU (30 μg/l) and photographed.
Colony formation assay
Equal numbers of cells silencing the expression of IRF-2 (IRF-2 siRNA1#, 2#) and control cells were seeded in 12-well plates. After 2 weeks, the colonies were stained with 0.1% crystal violet solution and photographed.
Soft agar assay
For base agar, 1% agar (DNA grade) was melted in microwave and cooled to 40°C in a water bath, and 2× DMEM/F12 + additives were warmed to 40°C in water bath followed by mixing equal volumes of the two solutions to give 0.5% agar + 1× DMEM/F12 + additives. Then, 1.5 ml was added to Petri dish. For top agar, 0.7% agar (DNA grade) was melted in microwave and cooled to 40°C in a water bath, and 2 DMEM/F12 + additives were warmed to the same temperature. Cells were trypsinized, counted, and added 0.1 ml of cell suspension to 10-ml centrifuge tubes. We labeled 35-mm Petri dishes with base agar appropriately. For top agar, 3 ml 2× DMEM/F12 + additives was added, 3 ml 0.7% agar was stored to a tube and mixed gently, and 1.5 ml to each replicate plate was added. Assay was incubated at 37°C in humidified incubator for 10–14 days, and colonies were counted using a dissecting microscope. All the experiments were repeated at least three times using triplicate plates.
Apoptosis measurement by flow cytometry
Cells were trypsinized, washed twice with PBS, and fixed in ice-cold 70% ethanol (4°C, overnight). After treatment with RNase A (200 μg/ml) for 1 h at 37°C, the cells were stained with propidium iodide (20 μg/ml, Sigma) for 1 h in dark at 37°C. The index of apoptosis was analyzed by flow cytometry.
Statistical analysis
Wilcoxon test was used to evaluate the difference between protein levels of IRF-2 in cancer samples and matched normal pancreatic tissues. Chi-square test was used to assess the associations between IRF-2 expression status and clinicopathological factors. Student’s t test was used to analyze the difference between groups. Univariate survival analysis was performed by using the Kaplan–Meier method and then analyzed by the log-rank test. p value of less than 0.05 was considered to be statistically significant. The data were analyzed by using the SPSS 16.0 statistical software for Windows (SPSS, Chicago, IL, USA).
Results
IRF-2 was up-regulated in pancreatic cancer
To investigate the potential role IRF-2 in pancreatic cancer, the expression of IRF-2 was analyzed in tumor samples of pancreatic cancer. We first examined the protein levels of IRF-2 in tumor species by immunohistochemistry using tissue array (Fig. 1a). It was found that the expression of IRF-2 was elevated in tumor samples compared with the paired normal tissues. As indicated in Fig. 1a, the expression of IRF-2 was dominant in the nucleus of the pancreatic cancer cells, which was similar to the previous studies [26]. We further confirmed this result using real-time PCR to examine the mRNA level and western blot analysis to detect the protein level of IRF-2 in tumor species and the paired normal tissues (Fig. 1b, c). These results implied that IRF-2 might exert an important role in pancreatic cancer.

The expression of IRF-2 was up-regulated in pancreatic cancer. a Immunohistochemical analysis of IRF-2 expression in pancreatic cancer and paired normal tissues. b Relative expression of IRF-2 mRNA in paired human pancreatic cancer samples and normal pancreatic tissues. Real-time PCR was performed on 30 paired pancreatic cancer samples. Data were calculated from triplicates. c The expression of IRF-2 protein level in pancreatic tumors. IRF-2 was examined in seven randomly selected paired pancreatic samples by western blot
The expression of IRF-2 was associated with the clinical features of patients with pancreatic cancer and with the patients’ survival
We next examined the correlation between the expression of IRF-2 and the clinical features of the patients. It was found that the expression of IRF-2 was correlated with tumor size, differentiation, and TNM stages. However, gender, ages, tumor location, nerve invasion, vessel invasion, and lymph node metastasis showed no correlation with the expression of IRF-2 (Table 1).
In the follow-up studies, the overall median survival time of the patients was 14.2 months. The Kaplan–Meier univariate analysis using the log-rank test revealed that patients with high expression of IRF-2 displayed significantly reduced median overall survival compared to patients with low expression of IRF-2 (high expression IRF-2: median overall survival 11.5 months; low expression IRF-2: median overall survival 16.5 months, p = 0.027) (Fig. 2). Furthermore, it also suggested that lymph node metastasis (p = 0.049) and TNM stage (p < 0.001) had significant correlation with the survival.

Over-expression of IRF-2 dictated poor survival in Kaplan–Meier survival durations analysis in pancreatic cancer. Kaplan–Meier curves of survival durations showed the correlation between IRF-2 expression and the overall survival of the patients
Silencing the expression of IRF-2 inhibited the growth of pancreatic cancer cells
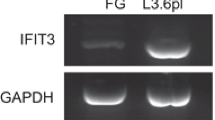
To examine the effects of IRF-2 on the growth of pancreatic cancer cells, the expression of IRF-2 was knocked down in MIAPaCa-2 and PANC-1 cells by two RNAi sequence (si 1# and si 2#) (Fig. 3a). The growth of MIAPaCa-2 and PANC-1cells was examined by MTT assay (Fig. 3b). Obviously, knockdown on the expression of IRF-2 inhibited the growth of the pancreatic cancer cells but not cell migration (Figs. 3b and S1). The BrdU incorporation assay and crystal violet assay (Fig. 3c, d) further confirmed this result. In addition, we determined the effects of IRF-2 knockdown on the colony forming of MIAPaCa-2 cells in soft agar, an anchorage-independent growth ability of cancer cells. As shown in Fig. 3e, f, down-regulation of IRF-2 reduced the number of colonies formed in soft agar. Collectively, these data indicated that IRF-2 is involved in the growth of pancreatic cancer cell and implicated its role in tumorigenesis.

Knockdown of IRF-2 inhibited the growth of pancreatic cancer cells. a Knockdown on the expression of IRF-2 in MIAPaCa-2 cells and PANC-1 cells. b Effects of IRF-2 knockdown on growth in MIAPaCa-2 cells and PANC-1 cells. The cell proliferation rate was examined by MTT assay. Results represent the mean ± SD of three experiments. c Effects of IRF-2 knockdown on cell proliferation in MIAPaCa-2 cells. The proliferation was measured by BrdU incorporation assay. d Crystal violet assay. Colonies were fixed, stained, and photographed with an inverted phase contrast microscope. e Soft agar assay. MIAPaCa-2 cells stably transfected with plasmids expressing indicated IRF-2 siRNA or control siRNA were incubated for 14 days in soft agar. The photos indicated colonies at 14th day of soft agar assay. f Colonies in soft agar were calculated. The results represent the mean ± SD of three independent experiments. *p < 0.05
Knockdown on the expression of IRF-2 regulated the expression of genes related to proliferation and apoptosis
IRF-2 was reported to regulate the expression of several genes which are implicated in cell proliferation and apoptosis [26]. Therefore, the effects of knockdown IRF-2 on the expression of related molecules were examined. In MIAPaCa-2 and PANC-1 cells, knockdown on the expression of IRF-2 up-regulated the expression of PARP and BAX and cleaved caspase-8 (Fig. 4a), all of which are pro-apoptosis gene [28]. On the other hand, knockdown on the expression of IRF-2 in the cells down-regulated the proliferation-related genes, cyclin D1 and PCNA (Fig. 4b). Furthermore, we detected the expression of PARP and BAX in five tumor samples with different IRF-2 expression level (Fig. 4c). The results suggested that the expression of PARP and BAX was inversely correlated with IRF-2.

Effects of IRF-2 knockdown on the expression of proliferation- and apoptosis-related molecules. a In MIAPaCa-2 cells and PANC-1 cells, IRF-2 knockdown up-regulated the expression of molecules related to apoptosis. b In MIAPaCa-2 cells and PANC-1 cells, IRF-2 knockdown regulated the expression of molecules related to proliferation. c The expression of IRF-2, PARP, and BAX protein in five pancreatic primary tumor samples was analyzed by western blot. d Apoptosis in MIAPaCa-2 cells was measured using FACS on flow cytometry. Silencing the expression of IRF-2 sensitized MIAPaCa-2 cells to apoptosis
In addition, as shown in Fig. 4d, silencing the expression of IRF-2 sensitized MIAPaCa-2 cells to apoptosis. These results suggested that IRF-2 exerted the oncogenic function on pancreatic cancer cell through regulating cell proliferation and apoptosis [29, 30].
Discussion
As a common malignancy, pancreatic cancer mostly originates from duct epithelial cells [1, 2, 29]. The development and progression of pancreatic cancer could be divided into several stages according to specific histological traits, which suggest the complicated cause of this disease [7]. Thus, the function of new genes which might play oncogenic or tumor suppressive roles needed to be identified in pancreatic cancer.
In our study, we first characterized the protein expression level of IRF-2 in paraffin-embedded samples using tissue array. It revealed that the expression of IRF-2 was higher in tumors than the paired normal samples. Real-time PCR and western blot also confirmed the result. Further, the statistic analysis revealed the correlation of IRF-2 and the clinicopathologic characteristics (Table 1), which illustrated the possible role of IRF-2 in the prognosis of pancreatic cancer. Kaplan–Meier survival curves showed that the patients with low IRF-2 expression had significantly higher cumulative survival rates compared to the patients with high IRF-2 expression (Fig. 2), which revealed that IRF-2 would be an important role in histological trait and prognosis of pancreatic cancer.
IRF-2 is an interferon regulatory factor and has been known to oppose the activity of IRF-1, a potential tumor suppressor [16, 18, 31, 32]. In diverse types of cancers, the expression of IRF-2 was found to positively associate with the malignant phenotype and be critical for the tumorigenicity of cancer cells [20, 24, 26]. Previous studies indicated that IRF-2 exerts it oncogenic activity in ESCC by affecting expression of certain genes involved in cell proliferation and apoptosis [25, 26]. In this study, the up-regulation of IRF-2 was observed in the tumor samples and would be important for the development of pancreatic cancer; in the meantime, the expression of IRF-1 and IRF-3 was dramatically down-regulated in pancreatic cancer (Fig. S2). Also molecules related to proliferation and apoptosis were examined, and decreased expression of cyclin D1 and PCNA by IRF-2 knockdown was observed (Fig. 4b). Cyclin D1 is the promoter of cell cycle and also contributes to tumorigenesis of pancreatic cancer [9, 30]. Therefore, in pancreatic cancer cell, cyclin D1 seems to be a downstream factor of IRF-2 to promote cell proliferation. Meanwhile, IRF-2 knockdown also led to up-regulation of PARP, BAX, and activation of caspase-8 (Fig. 4a), which are involved in cell apoptosis and have been known to inhibit development of pancreatic cancer [28, 33, 34]. In addition, the expression of PARP and BAX in pancreatic tumors was inversely correlated with that of IRF-2 (Fig. 4c). These results illustrated that IRF-2 could facilitate cell growth of pancreatic cancer cell by inhibiting cell apoptosis besides accelerating cell proliferation and suggested the oncogenic role of IRF-2 in pancreatic cancer cell.
However, based on these interesting findings, it should be noted that the nuclear/cytoplasmic localization of IRF-2 and the direct effectors of IRF-2 involved in these effects are worthy of further investigation in order to find more specific therapeutic targets of pancreatic cancer. Moreover, IRF-1 which has interaction with IRF-2 acts as a tumor suppressor and causes apoptosis in cancer cells [31, 32], and the relationship of IRF-1 and IRF-2 in the progression of pancreatic cancer should be further investigated.
In summary, our study suggested that IRF-2 was highly expressed in pancreatic cancer and was inversely associated with the overall survival of patients. IRF-2 played the important role for the growth of pancreatic cancer cells. At the same time, these results help explain why high expression of IRF-2 is observed in pancreatic cancer samples and implied that down-regulation of IRF-2 would be a new treatment target for pancreatic cancer
References
Warshaw AL, Fernandez-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;316:455–65.
Hidalgo M. Pancreatic cancer. N Engl J Med. 1992;362:1605–17.
Maiello E. A phase II trial of etoposide, folinic acid, fluorouracil and epirubicin in advanced pancreatic carcinoma. Clin Ter. 1998;149:351–5.
Holzman DC. Pancreatic cancer: will incremental advances begin to make a difference? J Natl Cancer Inst. 1996;102:1821–3.
Costello E, Neoptolemos JP. Pancreatic cancer in 2010: new insights for early intervention and detection. Nat Rev Gastroenterol Hepatol. 2010;8:71–3.
Milandri C, et al. Intra-arterial chemotherapy of advanced pancreatic cancer: a single center experience. Hepatogastroenterology. 2007;54:2373–7.
Hruban RH, et al. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–72.
Garcea G, et al. Molecular prognostic markers in pancreatic cancer: a systematic review. Eur J Cancern. 2005;41:2213–36.
Saif MW, Karapanagiotou L, Syrigos K. Genetic alterations in pancreatic cancer. World J Gastroenterol. 2007;13:4423–30.
Ghiorzo P, et al. INK4/ARF germline alterations in pancreatic cancer patients. Ann Oncol. 2004;15:70–8.
Naccarati A, et al. Genotype and haplotype analysis of TP53 gene and the risk of pancreatic cancer: an association study in the Czech Republic. Carcinogenesis. 2009;30:666–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Harada H, Taniguchi T, Tanaka N. The role of interferon regulatory factors in the interferon system and cell growth control. Biochimie. 1998;80:641–50.
Watanabe N, Taniguchi T. Involvement of positive (IRF-1) and negative (IRF-2) transcription factors in the gene regulation of the type I interferon system. Tanpakushitsu Kakusan Koso. 1992;37:2813–22.
Nguyen H, Hiscott J, Pitha PM. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997;8:293–302.
Harada H, et al. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science. 1993;259:971–4.
Xi H, et al. Co-occupancy of the interferon regulatory element of the class II transactivator (CIITA) type IV promoter by interferon regulatory factors 1 and 2. Oncogene. 1999;18:5889–903.
Lengyel P. Tumor-suppressor genes: news about the interferon connection. Proc Natl Acad Sci USA. 1993;90:5893–5.
Taniguchi T. Transcription factors IRF-1 and IRF-2: linking the immune responses and tumor suppression. J Cell Physiol. 1997;173:128–30.
Choo A, et al. siRNA targeting the IRF-2 transcription factor inhibits leukaemic cell growth. Int J Oncol. 2008;32:175–83.
Choo A, et al. The role of IRF-1 and IRF-2 transcription factors in leukaemogenesis. Curr Gene Ther. 2006;6:543–50.
Doherty GM, et al. Interferon regulatory factor expression in human breast cancer. Ann Surg. 2001;232:623–9.
Connett JM, et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 expression in breast cancer tissue microarrays. J Interferon Cytokine Res. 2005;25:587–94.
Nicolini A, Carpi A, Rossi G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006;17:315–37.
Wang Y, et al. Negative feedback regulation of IFN-gamma pathway by IFN regulatory factor 2 in esophageal cancers. Cancer Res. 2008;68:1136–43.
Wang Y, et al. Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer Res. 2007;67:2535–43.
Xi H, Blanck G. Interferon regulatory factor-2 point mutations in human pancreatic tumors. Int J Cancer. 2000;87:803–8.
Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–6.
Stanger BZ, Dor Y. Dissecting the cellular origins of pancreatic cancer. Cell Cycle. 2006;5:43–6.
Chung DC. Cyclin D1 in human neuroendocrine: tumorigenesis. Ann NY Acad Sci. 2004;1014:209–17.
Gao J, et al. IRF-1 transcriptionally up-regulates PUMA which mediates the mitochondrial apoptotic pathway in IRF-1 induced apoptosis in cancer cells. Cell Death Differ. 2010;17:699–709.
Kim EJ, et al. Interferon regulatory factor-1 mediates interferon-gamma-induced apoptosis in ovarian carcinoma cells. J Cell Biochem. 2002;85:369–80.
Hamacher R, et al. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64.
Westphal S, Kalthoff H. Apoptosis: targets in pancreatic cancer. Mol Cancer. 2003;2:6.
Acknowledgments
The authors gratefully thank the scholarship of SA-SIBS. This project is supported by China Postdoctoral Science Foundation (20100480636) to Yuezhen Deng.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Lei Cui, Yuezhen Deng, and Yefei Rong contribute equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Knockdown on the expression of IRF-2 did not affect cell migration. A Si con, si 1#, and si 2# MIAPaCa-2 cells were tested with the Boyden Chamber assay. After 15 h, cells that migrated through the filter membrane of the Boyden chamber were stained, and representative results are shown in the upper panel. B Quantification of migratory cells was analyzed with Image Pro software and is shown in the basal panel. (JPEG 85 kb)
Fig. S2
Down-regulation of IRF-1 and IRF-3 in pancreatic cancer. A Relative expression of IRF-1 mRNA in 30 pancreatic cancer tissues compared to the paired normal tissues. B Relative expression of IRF-3 mRNA in 30 pancreatic cancer tissues compared to the paired normal tissues. (JPEG 37 kb)
Rights and permissions
About this article
Cite this article
Cui, L., Deng, Y., Rong, Y. et al. IRF-2 is over-expressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biol. 33, 247–255 (2012). https://doi.org/10.1007/s13277-011-0273-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-011-0273-3