
Abstract
The genetic relationships of mantis shrimp Oratosquilla oratoria between the coastal waters of China and Japan were not well studied. To reveal the genetic differentiation and genetic structure among populations, we collected populations of mantis shrimp O. oratoria from the coastal waters of China and Japan to analyze the mtDNA control region variation. A total of 309 individuals of O. oratoria were collected from 13 localities (11 from China and 2 from Japan) and a segment of mitochondrial DNA control region was sequenced. Three hundred nine haplotypes were defined, yielding a very high haplotype diversity and nucleotide diversity. Two lineages of O. oratoria were revealed and displayed strong differences in the geographical distribution. In the coastal waters of China, the geographic distribution of the two lineages was completely separated by the Yangtze River estuary; however, the lineages showed geographic sympatry in two populations from Japan. Based on the lineage distribution, three groups were defined. There was no significant genetic differentiation among the populations within the three groups, indicating high gene flow within each group. Significant and negative values for Tajima D and Fu’s Fs tests, and mismatch distributions for two lineages indicated population expansion. The present result confirmed that the freshwater outflow from the Yangtze River formed a physical barrier and affected gene exchange. The different distribution patterns of the two lineages in coastal waters of China and Japan indicated that the larvae of O. oratoria were transferred from China to the coastal waters of Japan with a one-way gene flow.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fishery management is necessary both to ensure that the current fisheries can continue to be exploited in perpetuity and to assist in the recovery of depleted stocks (Ward 2000). Determining the population genetic structure can provide essential information to support resource recovery and to aid in delineating and monitoring populations for fishery management (Roldán et al. 2000). Molecular genetic techniques offer the ability to identify and elucidate the fishery stock structure, which may not be apparent from phenotypic or behavioral characteristics (Zhang et al. 2006).
The mantis shrimp Oratosquilla oratoria belonging to the genus Oratosquilla (Crustacea: Stomatopoda: Squillidae) is widely distributed in the coastal waters of China and Japan (Hamano 1988). It is an important fishery species in the coastal area of China and is the major catch in the Bohai Sea and Yellow Sea (Du et al. 2016). Because of overfishing in recent years, the resources of O. oratoria were gradually reduced, which might influence the genetic diversity of populations. For the effective conservation and exploitation of O. oratoria, it is necessary to obtain better knowledge of its genetic background. At present, there are several studies on the genetic diversity of O. oratoria. For example, significant genetic differentiation among the four populations of O. oratoria was found based on the mitochondrial COI gene (Zhang et al. 2016; Du et al. 2016). Two genetically differentiated groups of O. oratoria were identified in the coastal waters of China that had the Yangtze River Estuary as the dividing line (Du et al. 2016). However, there was a lack of genetic structure within the two groups. Two reasons may explain the lack of genetic structure within these two groups. The first reason was that the mtDNA COI and 16S rRNA, with a slower rate of evolution, is unable to reveal the weaker genetic structure. The second reason was that there was no genetic structure among the populations within geographic groups. To further investigate the genetic structure within the two lineages, it is necessary to select more effective DNA markers. The mtDNA control region is the most polymorphic region in the mtDNA genome (Stoneking et al. 1991). Based on the rapid rate of evolution in mtDNA (Hoelzel et al. 1991), the mtDNA control region is particularly suitable for population genetic studies (Liu et al. 2007). A previous genetic study using the control region of O. oratoria in Hong Kong revealed a genetic structure among populations (Lui et al. 2010). Additionally, the genetic relationship of O. oratoria from coastal waters of China and Japan was also unknown because the previous studies had no samples from the coastal waters of Japan.
To reveal the genetic differentiation and genetic structure among populations, we collected populations of O. oratoria from the coastal waters of China and Japan to analyze the mtDNA control region variation. The following questions are addressed: (1) the genetic relationships of O. oratoria between the coastal waters of China and Japan and (2) the genetic structure of O. oratoria among populations.
Materials and methods
Sample collection
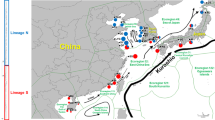
From 2011 to 2016, three hundred nine individual O. oratoria were collected from 13 localities in the coastal waters of China and Japan (11 from China and 2 from Japan) (Fig. 1; Table 1). Muscle samples were obtained and preserved in 95% ethanol or frozen for subsequent DNA extraction.

Map of the study area depicting sample locations and a schematic map of currents. Populations are marked by abbreviations that correspond to Table 1 (1. Kuroshio Current, 2. Taiwan warm current, 3. Tsushima Current, 4. Yellow Sea Warm Current, 5. Bohai Gulf Circulation, 6. Yellow Sea Coastal Current, 7. Lunan Coastal Current, 8. China coastal Current)
DNA extraction, amplification, and sequencing
Genomic DNA was isolated from muscle tissue by proteinase K digestion followed by a standard phenol–chloroform method (Sambrook et al. 1989). The DNA was subsequently resuspended in 100 µL of TE buffer (10 mmol/L Tris–Cl, 1 mmol/EDTA, pH = 8.0).
A portion of the mtDNA control region was amplified with the following primers: OO-F1 (5′-TCAAATAGAAAACAAATAGCCAG-3′) and OO-F2 (5′-CATAATTTATCCTATCAAGATAATC-3′), which were specifically designed for this study using the software Primer Premier 5.0 (Singh et al. 1998). The amplification reaction was carried out in 50 µL volumes containing 1.25U Taq DNA polymerase (Takara Co., China), 20 ng template DNA, 200 nmol/L forward and reverse primers, 200 µmol/L of each dNTPs, 10 mmol/L Tris, pH 8.3, 50 mmol/L KCl, 1.5 mmol/L MgCl2. The PCR amplification was performed in a thermal cycler under the following conditions: 4 min initial denaturation at 94 °C and 35 cycles of 1 min at 94 °C for denaturation, 1 min at 53 °C for annealing, and 1 min at 72 °C for extension, and a final extension at 72 °C for 10 min. All sets of PCRs included a negative control reaction tube in which all reagents were included, except the template DNA. PCR product was purified with Gel Extraction Mini Kit (Watson BioTechnologies Inc., Shanghai). The purified product was used as the template DNA for cycle sequencing reactions performed using BigDye Terminator Cycle Sequencing Kit (ver. 2.0, PE Biosystems, Foster City, California), and sequencing was conducted on an ABI Prism 3730 (Applied Biosystems) automatic sequencer with both forward and reverse primers. The primers used for sequencing were the same as those for PCR amplification.
Data analysis
Sequences were edited using the software SeqMan in DNAStar packages (http://www.dnastar.com/t-dnastar-lasergene.aspx). Molecular diversity indices, such as the numbers of haplotypes, polymorphic sites, transitions, transversions and indels, were obtained using Arlequin v 3.5 (Excoffier and Lischer 2010). Genetic variation was assessed using two estimators: nucleotide diversity (π) and haplotype diversity (h).
Genetic relationships among individuals were reconstructed using the neighbor-joining method (Saitou and Nei 1987) implemented in MEGA 6.0 (Tamura et al. 2013). Genetic distances were calculated by the Kimura two-parameter model (K-2P) (Kimura 1980), which was selected by the Software MEGA6.0. We used a bootstrap analysis with 1000 replicates to evaluate support for phylogenetic relationships. The genetic structure was investigated using analysis of molecular variance (AMOVA) in Arlequin. To test the genetic relationship of O. oratoria between the coastal waters of China and Japan, we tested with AMOVA a three-group structure. Based on the frequency of lineages, samples from Tangshan, Dongying, Qingdao, Longkou and Lianyungang were placed in the north group. Samples from Zhoushan, Qixingliedao, Xiaoyangshan, Zhanjiang, Fangchenggang and Beihai were placed in the south group. Samples from Hokkaido and Hyogo were placed in the Japan group.
Two different approaches in Arlequin were applied to study the demographic changes in O. oratoria. First, the D test of Tajima and Fs test of Fu were used to test whether neutrality held (Tajima 1989; Fu 1997). Historic demographic expansions were also investigated to examine the frequency distributions of pairwise differences between sequences (mismatch distribution) (Rogers and Harpending 1992). Both the mismatch analysis and neutrality tests were performed in Arlequin. The concordance of the observed with the expected distribution under the sudden expansion model of Rogers was tested by means of a least squares approach (Rogers and Harpending 1992). Various expansion parameters (θ0, θ1 and τ) were estimated by a general nonlinear least-squares approach. The values of τ were transformed to estimates of real time since expansion with the equation τ = 2ut, where u is the mutation rate for the whole sequence under study and t is the time since expansion.
The divergence rate of 19% per million years (My) for control region of brown shrimp Farfantepenaeus aztecus and white shrimp Litopenaeus setiferus was applied to calculate the divergence time between the two lineages and expansion time within lineages in the present study (McMillen-Jackson and Bert 2003).
Results
An alignment of 840–865 bp from a portion of the mtDNA control region was analyzed for 309 individuals from 13 localities. A total of 308 haplotypes were defined by 490 polymorphic sites with 422 transitions, 137 transversions and 105 indels (Table 1). These polymorphisms defined high values, with a haplotype diversity of 1.0000 ± 0.0003 and a nucleotide diversity of 0.1720 ± 0.0817 (Table 1). Every individual corresponds to a haplotype, except that Hokkaido has two individuals that share a haplotype. The lowest haplotype diversity (h) was 0.9967 ± 0.0125 (Hokkaido), and the nucleotide diversity ranged from 0.0237 ± 0.0121 (Dongying) to 0.1506 ± 0.0751 (Hyogo). All haplotype sequences were deposited in GenBank, with accession number MG912104-MG912412.
The NJ tree was constructed using the 309 individuals and identified two lineages (labeled A and B; Fig. 2). The average net genetic distances (Kimura-2P model) between the lineages were 20.4%. Applying the molecular clock of control region (19%/Myr), the divergence time of lineages A and B was about 1.07 million years before present. There are obvious differences in the geographical distribution of the frequency of the two haplotype lineages (Fig. 3). All individuals from the Bohai Sea and Yellow Sea belonged to lineage A, and lineage B included individuals from the East China Sea and South China Sea. Two lineages coexisted in Hokkaido and Hyogo, although most of the individuals from Hokkaido and Hyogo existed in lineage A.

Neighbor-joining tree of individuals constructed using the K-2P distance for Oratosquilla oratoria. Bootstrap supports of > 90% in 1000 replicates are shown

Haplotype frequencies for Oratosquilla oratoria from the coastal waters of China and Japan. The area of the circle is proportional to the sample size
The genetic structure of the population of O. oratoria was investigated by AMOVA. When the thirteen populations were placed in one-group, the results indicated that most of the variance was explained among populations (72.06%). In contrast, dividing the 13 populations into three groups showed that the genetic variation among populations within the groups was negative (− 0.23) (Table 2). When the thirteen populations were placed in three groups, the results indicated that no significant genetic differentiation was found among populations within the north group, the south groups and the Japan group (Table 2). On the contrary, FST values indicated a strong and significant differentiation among groups (Table 3).
Pairwise FST values within the three groups were observed, ranging from − 0.0133 to 0.0268 (north group), − 0.0175 to 0.0067 (south group) and − 0.0388 (Japan group). The low pairwise FST values revealed no significant genetic structure, suggesting extensive gene flow within the three groups. The genetic differentiation among populations showed the highest FST values between lineage A and lineage B and ranged from 0.8957 to 0.9069 (Table 3). The largest genetic differentiation occurred between Dongying and Beihai.
The mismatch distributions of lineage A and lineage B were unimodal (Fig. 4) and roughly fitted to the expected distributions under the sudden expansion model. The tau value (τ), which reflects the location of the mismatch distribution crest, provides a rough estimate of the time when rapid population range expansion started. The observed values of the sudden expansion parameter (τ) were 18.3 U and 27 U of mutational time for lineage A and lineage B, respectively (Table 4). Estimate of time of expansion for lineage A, based on the rates mentioned above for control region, was 105,900 years ago. For lineage B, this estimate was 156,000 years ago. Tajima’s D and Fu’s Fs (Table 5) were used to verify the neutrality of O. oratoria populations. Significant and negative values of Tajima D and Fu’s Fs tests indicated population expansion.

The observed pairwise difference (bars) and expected mismatch distributions (solid line) of the mtDNA control region haplotypes in Oratosquilla oratoria
Discussion
We analyzed a portion of the mtDNA control region among O. oratoria populations, and our results supported the occurrence of two lineages, which was consistent with a previous mtDNA COI study (Du et al. 2016). However, a very interesting result was that the two lineages of O. oratoria coexisted in the Hokkaido and Hyogo populations.
Among China populations, O. oratoria revealed complete genetic break on both sides of the Yangtze River Estuary, which indicated existence of contemporary physical barrier to preventing the mixture of two lineages. The freshwater outflow from the Yangtze River was the possible candidate physical barrier that affected the genetic patterns of species. The genetic connection among geographic populations of benthic organism usually depends on the dispersion of planktonic larvae. Flow from the Yangtze River, the third largest river in the world with an average annual discharge of 8,961,011 m3, can influence surrounding salinity (Dong et al. 2012). Additionally, the larvae of O. ratoria were sensitive to low salinity and almost stopped feeding at a salinity of lower than 15 (Liu et al. 2006, 2012). Low salinity and north shift of discharge from the Yangtze River during the reproductive season of O. ratoria may cause a disconnection between the Yellow Sea populations and the East China Sea populations, which prevented the gene flow between the Yellow Sea and East China Sea. Another evidence was showed in the previous genetic study of O. ratoria in Hong Kong, revealing genetic heterogeneity among populations over a small spatial scale (Lui et al. 2010). Low salinities (average salinity: 10–20) caused by the summer discharge of freshwater from the Pearl River was responsible for the genetic difference between western and eastern waters of Hong Kong.
Similar to the present study, the barrier effect of the Yangtze River outflow was also reported to the limpet Cellana toreuma (Dong et al. 2012), the cocktail shrimp Trachypenaeus curvirostris (Han et al. 2015a) and Octopus ocellatus (Lü et al. 2009). However, it had no effect on gene exchange between populations of crab Charybdis japonica (Han et al. 2015b). Therefore, the Yangtze River outflow seemed to affect some, but not all, invertebrate species, depending on the differences in salt-tolerance ability.
The present study found that the two lineages of O. oratoria coexisted in the coastal waters of Japan and that no significant difference in lineage frequency occurred between the populations. The distribution of the two lineages may be caused by the one-way transport of larvae from the coastal waters of China to the Japanese Islands. Such a phenomenon was also indicated in a previous study, in which one-way gene flow affected the genetic variation in the population of Sebastes schlegeli (Yokota and Watanabe 1997). Under the effect of the Kuroshio Current and China Coastal Current, larvae of O. oratoria were transported from coastal waters of China into the coastal waters of Japan. This finding suggests that the one-way dispersal of marine organisms may play an important role in the genetic structure of species. However, the high frequency of lineage A in two Japanese populations (72%, 71.4%) showed more efficient dispersal of lineage A from northern coastal waters of China to Japanese coastal waters through coastline than the ocean current transportation of lineage B from southern populations of China to Japanese populations.
In general, O. oratoria showed an overall pattern of north–south differentiation, but no genetic structure within the three groups was detected. Marine species with broad dispersal ability during the planktonic stage usually have genetic homogeneity in a large spatial range. As adults of O. oratoria have a benthic life-style, the gene exchange mainly occurs through larval dispersal. The larvae of O. oratoria have a long planktonic stage that lasted between 36 and 59 days (Hamano and Matsuura 1987), indicating strong dispersal ability. Similar to the biological characters of O. oratoria, Charybdis bimaculata had a high larva dispersal ability, and wide genetic homogeneity was detected in the Yellow Sea and coastal waters of Japan (Han et al. 2015c).
The ocean current system also facilitates the dispersal of larvae of O. oratoria within three geographic groups. Larvae of O. oratoria from Tangshan to Lianyungang in north group were connected by three local coastal currents (Yellow Sea Coastal Current, Lunan Coastal Current, and Bohai Gulf Circulation). China coastal Current and Tsushima Current can connect the distant geographic populations within south group and Japan group, respectively. The transportation by ocean currents within groups may be the reason for the lack of genetic structure in three groups. The present result was consistent with the findings of Zhang et al. (2010).
The sequence data were tested using both neutrality tests and mismatch distribution to explain the demographic history of O. oratoria. Significant and negative values of Tajima D and Fu’s Fs tests indicated population expansion. The mismatch distribution for two lineages was unimodal, which also supported population expansion. The population expansion of marine organism was common in the Northwestern Pacific. The population expansions and contractions were closely associated with Pleistocene-era environmental fluctuations. Fluctuations in sea level, water temperatures and sea ice caused by glacial cycles are believed to have had a major influence on species distributions and the population connectivity of marine species (Hewitt 2000). As one of the most extensive continental shelves in the Western Pacific, during Quaternary glacial cycles, the Yellow Sea and most of the East China Sea were exposed (Wang 1999).
In summary, the results of this study indicated the following: (1) two lineages of O. oratoria coexisted in Japan group; (2) there was a lack of genetic structure among populations within the three groups, which could be used as three management units; and (3) there was a one-way gene transport of O. oratoria from the coastal waters of China to Japan. To have a comprehensive understanding of the population genetic structure in O. oratoria, it is necessary to use SSR to verify our results.
References
Dong YW, Wang HS, Han GD, Ke CH, Zhan X, Tomoyuki N, Williams GA (2012) The impact of Yangtze River discharge, ocean currents and historical events on the biogeographic pattern of Cellana toreuma along the China coast. PLoS ONE 7:e36178
Du XW, Cai SS, Yu CG, Jiang XQ, Lin LS, Gao TX, Han ZQ (2016) Population genetic structure of mantis shrimps Oratosquilla oratoria: testing the barrier effect of the Yangtze River outflow. Biochem Syst Ecol 66:12–18
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Hamano T (1988) Mating behavior of Oratosquilla oratoria (de Haan, 1844) (Crustacea: stomatopoda). J Crustacean Biol 8:239–244
Hamano T, Matsuura S (1987) Egg size, duration of incubation, and larva development of the Japanese mantis shrimp in the laboratory. Nippon Suisan Gakk 53:23–39
Han ZQ, Zhu WB, Zheng W, Li PF, Shui BN (2015a) Significant genetic differentiation between the Yellow Sea and East China Sea populations of cocktail shrimp Trachypenaeus curvirostris revealed by the mitochondrial DNA COI gene. Biochem Syst Ecol 59:78–84
Han ZQ, Zheng W, Zhu WB, Yu CG, Shui BN, Gao TX (2015b) A barrier to gene flow in the Asian paddle crab, Charybdis japonica in the Yellow Sea. ICES J Mar Sci 72:1440–1448
Han ZQ, Zheng W, Chen GB, Shui BN, Liu SF, Zhuang ZM (2015c) Population genetic structure and larval dispersal strategy of portunid crab Charybdis bimaculata in Yellow sea and East China sea. Mitochondr DNA 26:402–408
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hoelzel AR, Hancock JM, Dover GA (1991) Evolution of the cetacean mitochondrial D-loop region. Mol Biol Evol 8:475–493
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Liu HY, Xu HL, Lin YJ (2006) The effect of salinity on survival and growth mantis shrimp (Oratosquilla oratoria) in Dalian coast. J Dalian Ocean Univ 21:180–183
Liu JX, Gao TX, Wu SF, Zhang YP (2007) Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol Evol 16:275–288
Liu HY, Wang DX, Jiang YS, Chen L (2012) The effects of salinity on survival and food intake in mantis shrimp Oratosquilla oratoria larvae. J Dalian Ocean Univ 27:311–314
Lü ZM, Li H, Wu CW, Fan ZJ, Zhang JS (2009) Population genetic structure of Octopus Ocellatus in China coastal water based on 16S rDNA sequence analysis. Oceanol Et Limnol Sin 40:330–337
Lui KKY, Leung PTY, Ng WC, Leung KMY (2010) Genetic variation of Oratosquilla oratoria (crustacea: stomatopoda) across Hong Kong waters elucidated by mitochondrial DNA control region sequences. J Mar Biol Assoc UK 90:623–631
McMillen-Jackson AL, Bert TM (2003) Disparate patterns of population genetic structure and population history in two sympatric penaeid species in the southeastern United States. Mol Evol 12:2895–2905
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Roldán MI, Perrotta RG, Cortey M, Pla C (2000) Molecular and morphologic approaches to discrimination of variability patterns in chub mackerel, Scomber japonicus. J Exp Mar Biol Ecol 253:63–74
Saitou N, Nei M (1987) The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Singh VK, Mangalam AK, Dwivedi S, Naik S (1998) Primer premier: program for design of degenerate primers from a protein sequence. Biotechniques 24:318–319
Stoneking M, Hedgecock D, Higuchi RG, Vigilant L, Erlich HA (1991) Population variation of human mtDNA control region sequences detected by enzymatic amplification and sequence-specific oligonucleotide probes. Am J Hum Genet 48:370–382
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Wang PX (1999) Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Mar Geol 156:5–39
Ward RD (2000) Genetics in fisheries management. Hidrobiología 420:191–201
Yokota M, Watanabe S (1997) One-way gene flow by stocking and its effects on a fish population. Fish Sci 63:539–541
Zhang JB, Cai ZP, Huang LM (2006) Population genetic structure of crimson snapper Lutjanus erythropterus in East Asia, revealed by analysis of the mitochondrial control region. ICES J Mar Sci 63:693–704
Zhang DZ, Ding G, Zhang HB, Tang BP (2010) Genetic diversity of Oratosquilla oratoria from different sea area of Bohai Bay by mitchondrial COI gene. Nanjing Norm Univ 33:80–83
Zhang DZ, Ding G, Ge BM, Liu QN, Zhang HB, Tang BP, Yang G (2016) Geographical distribution, dispersal and genetic divergence of the mantis shrimp Oratosquilla oratoria, (Stomatopoda: Squillidae) in China Sea. Biochem Syst Ecol 65:1–8
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0604902) and the National Natural Science Foundation of China (31472281).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yang Zhang declares that she does not have conflict of interest. Zhiqiang Han declares that he does not have conflict of interest. Tianxiang Gao declares that he does not have conflict of interest. Huilai Shi declares that he does not have conflict of interest.
Ethical approval
This article does not contain any studies with human subjects by any of the authors. The study was approved by the Animal Care and Use Committee of Zhejiang University, and the animal experiment throughout the study was conducted according to the Chinese Ministry of Science and Technology Guiding Directives for Humane Treatment of Laboratory Animals.
Rights and permissions
About this article

Cite this article
Zhang, Y., Han, Z., Gao, T. et al. Genetic structure analysis of mantis shrimp Oratosquilla oratoria based on mitochondrial DNA control region sequence. Genes Genom 40, 1001–1009 (2018). https://doi.org/10.1007/s13258-018-0707-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0707-z