
Abstract
Rhododendron molle G. Don occupies an important phylogenetic node in the genus rhododendron with unique yellow flower and medicinal functions. However, only limited genetic resources and their genome information are available for the generation of rhododendron flowers. The next generation sequencing technologies enables generation of genomic resources in a short time and at a minimal cost, and therefore provide a turning point for rhododendron research. Our goal is to use the genetic information to facilitate the relevant research on flowering and flower color formation in R. molle. In total, 66,026 unigenes were identified, among which 31,298 were annotated in the NCBI non-redundant protein database and 22,410 were annotated in the Swiss-Prot database. Of these annotated unigenes, 9490 and 18,680 unigenes were assigned to clusters of orthologous groups and gene ontology categories, respectively. A total of 7177 genes were mapped to 118 pathways using the Kyoto Encyclopedia of Genes and Genomes Pathway database. In addition, 8266 simple sequence repeats (SSRs) were detected, and these SSRs will undoubtedly benefit rhododendron breeding work. Metabolic pathway analysis revealed that 32 unigenes were predicted to be involved in carotenoid biosynthesis. Our transcriptome revealed 32 engines that encode key enzymes in the carotenoid biosynthesis pathway, including PSY, PDS, LCYB, LCYE, etc. The content of β-carotene was much higher than the other carotenoids throughout the flower development. It was consistent with the key genes expression level in the carotenoid biosynthesis pathway by the Illumina expression profile analysis and the qRT-PCR analysis. Our study identified genes associated with carotenoid biosynthesis in R. molle and provides a valuable resource for understanding the flowering and flower color formation mechanisms in R. molle.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhododendrons, which are commonly referred to as rosage, are decorative shrubs with beautiful flowers that are widespread around the world in countries characterized by mild climates (Chosson et al. 1998). While there is a remarkably broad range of rhododendron flower colors, such as red, pink, purple, and white, there is no yellow-flowered cultivar of evergreen rhododendron (Ureshino et al. 2016). A yellow-flowered evergreen rhododendron has been the dream and goal of many rhododendron breeders. Rhododendron molle is a deciduous Rhododendron species with unique yellow flowers. Variations in the morphological characteristics and pigments of wild evergreen rhododendron flowers have been studied in detail (Huyen et al. 2016; Mizuta et al. 2009; Nakatsuka et al. 2008). It has been reported that the flower color of Rhododendron japonicum with yellow-flower is attributed to the presence of a carotenoid (Miyajima et al. 2000). Carotenoids are 40-carbon isoprenoids with polyene chains, which are usually responsible for yellow-orange petal colors and play an important role in furnishing flowers and fruits with distinct colors that are designed to attract pollinators (Hirschberg 2001). During the past decade, nearly all of the genes involved in the carotenoid biosynthesis pathway have been identified and characterized (Pandurangaiah et al. 2016; Rezaei et al. 2016; Yang et al. 2016). The differential expression of these genes may lead to differences in petal color (Sagawa et al. 2016; Xin; Li et al. 2015). However, there are few genetic data concerning carotenoids of R. molle. Therefore, the study of transcrioptome sequencing will provide an important molecular basis for study of R. molle and understanding its flowering mechanisms.
In recent years, the number of EST data sets has rapidly increased (Han et al. 2017; Xia et al. 2017). However, rhododendrons have little data available currently in DNA or protein sequence databases. It hampers the molecular breeding research of rhododendron. Sequencing of large genome remains expensive, even using next generation sequencing technologies. On the contrary, de novo transcriptome sequencing has proved to be accurate and efficient with much higher throughput and relatively low cost, providing opportunities to discover massive expressed sequence data for model and non-model species, such as chrysanthemum (Wang et al. 2017), tea (Li et al. 2017), and lettuce (Zhang et al. 2016). Moreover, the transcriptome is a complete set of transcripts in a cell at a specific developmental stage, providing information on amino acid content of proteins, gene expression and gene regulation. Thus the transcriptome analysis is essential to interpret the functional elements of the genome and reveal the molecular constituents of cells and tissues (Liu et al. 2012; Wang et al. 2009). In addition, transcriptome sequencing can provide a simple and effective method to develop a large number of simple sequence repeats (SSRs) marker and improved the efficiency of molecular marker development in many non-model species (Chen et al. 2016; Guo et al. 2016; Wang et al. 2016). To our knowledge, this is the first report on the characterization of transcrioptome of R. mollle. Hence, the study of transcrioptome sequencing will provide an important molecular basis for study of R. molle and understanding its flowering mechanisms.
In the present study, we used the Illumina sequencing platform to conduct the transcriptome sequencing analysis of mixed RNA separately extracted from flowers at four developmental stages of R. molle flower. A total of 123,120 different transcripts and 66,026 unigenes were identified. Meanwhile, a large number of SSRs were determined. Particularly, the transcripts involved in carotenoid biosynthesis were sequenced, aiming to facilitate color-regulating mechanisms research in rhododendron.
Materials and methods
Plant materials and RNA extraction
Rhododendron molle was grown in the field in Nanjing (China). Samples of flowers at four developmental stages were collected in 2016 as follows: tight bud stage (S1), soft bud stage (S2), initial flowering stage (S3) and full flowering stage (S4). The samples were collected, frozen immediately in liquid nitrogen, and stored in − 80 °C for RNA extraction. Total RNA was extracted using the Quick RNA isolation kit (Bioteke Corporation, Beijing, China) following the manufacturer’s instructions. RNA quality and purity were checked using a Nanodrop 2000 spectrophotometer (Thermo, Wilmington, DE, USA) and Qubit RNA Assay Kit in Qubit 2.0 Fluorometer (Life Technologies, CA, USA). The RNA integrity was assessed using the RNA Nano 6000 Assay Kit of Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
cDNA library construction and sequencing
A pooled sample containing equal amounts of purified total RNA from each of the four samples was sent to Biomarker Technology Co. Ltd (Beijing, China). Construction of the libraries and the RNA-Seq were performed using the Illumina HisSeq 2500 platform. Firstly, mRNAs were isolated using oligo (dT) magnetic beads and fragmented into small pieces using a fragmentation buffer. First strand cDNA was reverse transcribed by random hexamers, followed by second strand cDNA synthesis. The double-stranded cDNA was subjected to an end repair process and the addition of single nucleotide A. Then paired-end adapters were ligated to the ends of the cDNA fragments. Suitable fragments were selected as templates for polymerase chain reaction amplification. Finally, the library preparations were sequenced on an Illumina HiSeq 2500 platform.
De novo transcriptome assembly
Clean reads were obtained by removing reads with adapters, reads with unknown nucleotides larger than 5% and low quality reads from raw data. Transcriptome de novo was carried out by program Trinity (Grabherr et al. 2011). All transcripts were clustered and the longest transcript in each cluster was defined as the unigene for functional annotation. The initial assembly quality was evaluated using the total number of contigs, mean contig length, maximum contig length and N50 (Martin and Wang 2011). The Trinity utility was used to predict ORFs of at least 100 amino acids from the reconstructed contigs.
Gene annotation and pathway assignments
All assembled unigenes were searched against NCBI non-redundant protein sequences (Nr) to determine the putative mRNA functions using the BLAST algorithm with an E-value lower than 10−5 (Altschul et al. 1997). GO (Gene ontology) terms were extracted from the best hits obtained from the BLASTX against Nr using the Blast2GO program (Götz et al. 2008). The sequences with corresponding enzyme commission (EC) numbers obtained from blast2GO were mapped to the Kyoto Enzyclopedia of Genes and Genomes (KEGG) Web server (Kanehisa et al. 2008). The WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) was used to obtain GO functional classifications. In addition, Unigenes were also aligned to KOG/COG (Clusters of Orthologous Groups of proteins) database, Nt (NCBI non-redundant nucleotide database) and Swiss-Prot to predict and classify possible functions of unigenes (Tatusov et al. 2001).
Extraction and measurement of carotenoids
The samples of flowers at four development stages were collected from five trees. And each stage contained five flowers with three replicates. The carotenoids were extracted and measured according to the previous study (Yan et al. 2015). Frozen flower petals (0.8 g) were homogenized in 4 mL of acetone, and extracted for 24 h away from light at 4 °C. Then the extracts were centrifuged at 16,000×g for 10 min at 4 °C, and filtered through a 0.22 µm filter for analysis in the Agilent 1100 series HPLC system (Agilent, USA). Samples (20 µL of extract) were analyzed using a YMC-C30 chromatographic column coupled with a UV detector at 450 nm, using methyl tertiary butyl ether and methyl alcohol (V/V = 30:70) as solvents: the flow rate was 1.0 mL min−1 and temperature was 25 °C. The lutein, zeaxanthin, α-carotene and β-carotene were quantified by comparing the peak areas.
Quantitative real-time PCR analysis
Quantitative real-time PCR (qRT-PCR) analysis was used to verify the expression levels of genes identified in sequencing. The RNA samples used for qRT-PCR assays were the same as the sequencing samples. Housekeeping gene actin was used as the control gene (Xiao et al. 2016). All primers are listed in Table S1.The qRT-PCR reactions were performed using a SYBR Premix Ex Taq Kit (TaKaRa, Dalian, China) following manufacturer’s instructions and was analyzed in the ABI 7500 Real-Time PCR system (Applied Biosystems, CA, USA). In a white 96-well plate (Axygen, CA, USA), 10 µL SYBR Master Mix and 0.4 µL of 10 µM forward primer and reverse primer was used in combination with 2 µL cDNA template, resulting in a total volume of 20 µL. Cycling conditions involved 10 s at 95 °C followed by 40 cycles of 5 s 95 °C and 34 s 60 °C. A melting dissociation curve was created to identify amplicon characteristics. The final threshold cycle values were average values. Each reaction was performed in triplicate. Analysis of the relative gene expression data was conducted using the 2−ΔΔCt method.
Development of SSR markers
Simple sequence repeats (SSRs) were detected using the MISA software (http://pgrc.ipk-gatersleben.de/misa/). The parameters were adjusted for identification of perfect di-, tri-, tetra-, penta-, and hexa-nucleotide motifs with a minimum of 6, 5, 5, 5 and 5 repeats, respectively.
Results
Sequencing and de novo assembly
A total of 20,143,879 (5.07 G data) clean reads were obtained after removing duplicate reads, trimming adapters and low quality sequences. The Q30 percentage and GC-content were 88.47 and 47.69%, respectively. The sequencing raw data were deposited in the NCBI database and can be accessed in the Short Reads Archive (SRR6056607).
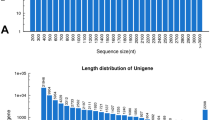
The trinity software generated 124,120 all-transcripts with length > 200 bp, mean length of 1093 bp and an N50 of 1862 bp (Table 1). 66,026 all-unigenes were achieved with an N50 of 1243 bp. There were 26,539 (40.19%) unigenes of 200–300 bp; 16,007 (24.24%) unigenes of 300–500 bp; 10,656 (16.14%) unigenes of 500–1000 bp; 8053 (12.20%) unigenes of 1000–2000 bp; and the remaining 4771 (7.23%) unigenes were > 2000 bp (Table 1). The length distribution of unigenes is shown in Fig. 1.

Length distribution of de novo assembled unigenes
Functional annotation and classification
All of the contigs generated by Illumina sequencing were aligned to public protein and gene databases. The overall functional annotation is shown in Table 2. The contigs were compared to the sequences in the NR database, using BLASTX (E-value < 1e−3). A total of 31,298 unigenes (47.40%) returned a significant BLASTX result. The contigs homologous to sequences in the NR database were annotated using Blast2GO. A total of 18,680 (28.29%) unigenes was assigned to 4658 GO term annotations, which could be classified into three categories: biological processes, molecular functions and cellular components (Fig. 2). Among biological processes, “oxidation–reduction process” represented the most abundant subcategory followed by “protein phosphorylation” and “regulation of transcription”. In the molecular function category, “ATP binding” and “Zinc ion binding” were the most represented GO terms. Regarding cellular components, the major GO terms were “plasma membrane” and “internal component of membrane”.

Gene ontology (GO) classification of assembled unigenes. The 66,026 matched unigenes were classified into three functional categories: biological process, cellular component and molecular function
The KOG analysis allowed the functional classification of 17,739 unigenes (Fig. 3). The most frequently identified classes were “general function” (4455, 25.11%), followed by “posttranslational modification” (1976, 11.14%) and “signal transduction mechanisms” (1723, 9.71%).

Clusters of eukaryotic orthologous groups (KOG) classification
KEGG pathway and predicted coding sequences (CDS)
A total of 7177 unigenes had significant matches in KEGG, mapped with 118 KEGG pathways (Table S2). Figure 4 summarizes the top 20 represented biological pathways. The most highly represented pathways were “Ribosome” (349 unigenes, 4.86%, ko03010) and “Oxidative phosphorylation” (268 unigenes, 3.73%, ko00190). Four pathways were found to be related to plant color: “Anthocyanin biosynthesis” (2 unigenes, ko00942), “Carotenoid biosynthesis” (32 unigenes, ko00906), “Flavone and flavonol biosynthesis” (7 unigenes, ko00944) and “Flavonoid biosynthesis” (38 unigenes, ko00941).

KEGG classification of unigenes
Based on the public protein database, a total of 55,456 CDSs was obtained. Among these CDSs, 47,695 were between 100 and 1000 bp, 5934 were between 1000 and 2000 bp, and 1827 were over 2000 bp (Fig. 5).

Length distribution of predicted CDS
Key enzymes related to the carotenoid pathway
There were 32 unigenes annotated as encoding enzymes involved in carotenoid synthesis based on the KEGG pathway assignments while only 2 unigenes encoding enzymes of the anthocyanin biosynthesis pathway were identified (Table S2). Phytoene synthase (PSY), a rate-limiting enzyme in the carotenoid biosynthesis pathway, catalyzes a head-to-head coupling of two geranylgeranyl diphosphate (GGPP) molecules to produce phytoene (Fig. 6). Then phytoene is catalyzed to lycopene by phytoene desaturase (PDS), ζ-carotene desaturase (ZDS) and carotenoid isomerase (CRTISO). All of these enzymes were identified: c18470.graph_c0 annotated as PSY, c25193.graph_c0 as PDS, c21632.graph_c0 as ZDS and c29096.graph_c0 as CRTISO. Then the carotenoid biosynthetic pathway was split into two routes. In one route, lycopene ε-cyclase (LCYE) and lycopene β-cyclase (LCYB) catalyzed the cyclization of lycopene to produce α-carotene, which was subsequently converted to lutein (Chen et al. 2015). Transcript c30693.graph_c0 and c25695.graph_c0 were annotated as LCYB and LCYE, respectively. Enzymes in the other route were essential for the biosynthesis of abscisic acid (ABA). The route yielded β-carotene with catalysis by LCYB. Then β-carotene was catalyzed by β-ring hydroxylase (CHYB, c18642.graph_c0), zeaxanthin epoxidase (ZEP, c12244.graph_c0), and violaxanthin de-epoxidase (VDE, c11312.graph_c0) produced zeaxanthin and violaxanthin. Finally, violaxanthin was converted to ABA by neoxanthin synthase (NSY) and 9-cis-epoxycarotenoid dioxygenase (NCED, c24238.graph_c0, c18704.graph_c0, c46847.graph_c0 and c62319.graph_c0).

Simplified pathways for the carotenoid biosynthesis. Numbers in the brackets indicated numbers of candidate unigenes annotated in our research
To study whether high carotenoid percentage results from high expression levels of carotenoid biosynthesis related genes in R. molle, the expression of transcripts encoding key enzymes for carotenoids was analyzed. Transcript c21632.graph_c0_seq1, annotated as Zeta-carotene desaturase (ZDS), reached the highest expression level (267.79 FPKM). PSY (C18470.graph_c0), PDS (c25193.graph_c0), CHYB (c18642.graph_c0) and ZEP (c12244.graph_c0) were 104.92 FPKM, 71.38 FPKM, 35.56 FPKM and 19.4 FPKM, respectively. Meanwhile, the expression of LCYB (c30693.graph_c0) was 26.01 FPKM, 3.22-fold higher than LCYE (c25695.graph_c0). These finds impel us to hypothesize that most of lycopenes are converted into β-carotenes by LCYB, and β-carotenes are further modified by hydroxylation or epoxidation, providing a variety of structural features.
Changes of carotenoid content during flower development
The results showed that there were a large number of carotenoids accumulated in the petal during the flower early development. The concentration ranges of lutein, zeaxanthin, α-carotene and β-carotene were 4.24–48.15 µg g−1 FW, 1.72–18.71 µg g−1 FW, 1.05–2.63 µg g−1 FW, 46.22–95.25 µg g−1 FW, respectively. The content of β-carotene was much higher than the other carotenoids during the flower development (Fig. 7). The α-carotene concentration remained low throughout flower development, and declined to the lowest at stage S3. The content of β-carotene tended to decline at stage S2 and rise to the highest at stage S4. On the contrary, the content of lutein and zeaxanthin tended to rise at stage S2 and decline to the lowest at stage S4. It was indicated that β-carotene played an important role in the coloring of R. molle flower.

Changes of carotenoids content during flower development in R. molle. Four flowers developmental stages were examined in this study as follows: tight bud stage (S1), soft bud stage (S2), initial flowering stage (S3), full flowering stage (S4). Error bars show the standard error calculated from three biological replicates
Validation of Illumina sequencing result by qRT-PCR
To confirm the accuracy and reproducibility of the Illumina expression profiles, qRT-PCR analysis was carried out to analyze the expression level of five genes related to carotenoid catabolism (Fig. 8). The expression levels of each gene were measured through qRT-PCR and compared with its abundance from sequencing data. The qRT-PCR analysis showed that PSY (C18470.graph_c0) and PDS (c25193.graph_c0) were highly expressed during the flower development, followed by CHYB (c18642.graph_c0), LCYB (c30693.graph_c0), and LCYE (c25695.graph_c0). It is generally agreed well with the sequencing result, thus confirmed the Illumina expression profile analysis.

Expression levels of five genes related to carotenoid catabolism throughout the flower development of R. molle. a Expression levels of PSY and PDS at different stages of flower development. b Expression levels of LCYB, LCYE and CHYB at different stages of flower development. Error bars show the standard error calculated from three biological replicates
SSR discovery
Approximately 3–7% of expressed genes in the plant genome contain putative SSR motifs, which are mainly located in the untranslated regions of mRNA (Liu et al. 2012). SSRs are widely used for genetics, evolution and breeding research. To explore SSR profiles in the R. molle, the assembled sequences (≥ 1 kb) were mined for SSRs. In total, 8266 SSRs were obtained from 12,824 unigenes with 1707 unigene sequences contained more than one SSR. The di-nucleotide repeat motif was the most abundant, accounting for 56.15%, followed by tri-nucleotide repeat motif (15.56%), tetra-nucleotide (0.57%), penta-nucleotide (0.17%) and hexa-nucleotide repeat units (0.08%) (Table 3).
The distribution and frequency of classified SSRs were analyzed (Fig. 9). Di-nucleotide was the most common repeat motif with a frequency of 56.15%, followed by tri-nucleotide (15.56%), tetra-nucleotide (0.57%), penta-nucleotide (0.17%), and hexa-nucleotide (0.08%). Among the dinucleotide repeats, the TC/GA (27.44%) and AG/CT (24.28%) were the most frequency motifs, accounting for 92.11% of the total dinucleotide repeats, much higher than that of Dendrocalamus latiflorus Munro and Huperzia serrate Thunb (Liu et al. 2012; Luo et al. 2010). Among the trinucleotide repeats, GAA/CTT (1.44%) and AGA/TCT (1.16%) were the most frequent motifs. The SSRs mining from this database will be valuable resources for genetic manipulations in R. molle.

Frequency distribution of SSRs based on motif types
Discussion
The high-throughput sequencing technology is an efficient and reliable tool for the transcriptome analysis in organisms without a reference genome. Prior to our work, there was no sequencing information on R. molle public database. The cDNA library was constructed using pooled RNA samples. In this study, a total of 5.07 Gb clean data was generated and assembled into 66,026, 31,542 of which were annotated in public databases, and 32 carotenoid structural genes was found. In addition, 8266 SSRs were identified from 12,824 unigenes. This study provides valuable information for future molecular research on R. molle.
In total, 7177 unigenes were assigned to different pathways in the KEGG classification system. Four pathways, flavone and flavonol biosynthesis (ko00944), flavonoid biosynthesis (ko00941), anthocyanin biosynthesis (ko00942), and carotenoid biosynthesis (ko00906), involved in the color formation of plant flower were identified. The carotenoid biosynthesis pathway is important for the synthesis of plant pigments. The color of yellow flowers is usually caused by the presence of carotenoids. The carotenoid metabolic pathway has been extensively studied (Yuan et al. 2015). In pale-yellow petals of marigold, expression levels of nearly all the carotenogentic genes are relatively lower than those in orange petals (Ohmiya 2013). In chrysanthemum petals, no significant difference in the expression levels of carotenoid biosynthesis genes was detected between the white and yellow petals. However, carotenoids are degraded into colorless compounded for CmCCD4a highly expressed in the white-flowered chrysanthemum, which results in the white petal color (Ohmiya et al. 2006). The yellow color fades in azalea petals are also contributed by high expression of CCD4 (Ureshino et al. 2016). Disruption of CCD4 gene can convert flower color from white to yellow in Brassica species (Zhang et al. 2015). White petals of chrysanthemum turned yellow after RNAi construct of CmCCD4a was introduced (Ohmiya et al. 2006). However, the mechanisms that control carotenoid catabolism in R. molle flower are far from conclusive. Therefore, the estimation of candidate enzymes in the biosynthesis of carotenoids will facilitate further research on the underlying mechanisms. In this study, 32 unigenes belonging to the carotenoid synthesis were discovered. According to the sequencing result, the most highly expressed genes related to carotenoid biosynthesis were ZDS, followed by PSY and PDS while no unigene related to CCD4 was detected. In addition, the qRT-PCR results showed that the expression levels of PSY and PDS were low during the early flowering stage, and increased as the flowers developed. The expression level of LCYB was lower than LCYE during the early flowering stage, but higher than LCYE during the later stages. This result was consistent with the changes of carotenoid content in the flower petals of R. molle during the flower development. The content of β-carotene was much higher than the other carotenoids during the flower development, which declined at the stage S2 and rose to the highest at stage S4. The α-carotene concentration remained low throughout flower development. The identification of enzymes and related genes involved in the carotenoid biosynthesis and metabolic pathway would facilitate both color-regulating mechanisms and molecular breeding research in rhododendron.
SSRs were developed as co-dominant and multi-allelic, powerful molecular markers for comparative genetic mapping and genotyping since they are co-dominant, multi-allelic, highly polymorphic, locus-specific, and transportable among species within genera (Bruni et al. 2012; Varshney et al. 2005a, b). Six SSRs have been successfully used to analyze the genetic structure of rhododendron hybrid population for better characterize the evolution of hybridization events (Bruni et al. 2012; Gismondi and Canini 2013). However, only a few SSRs were developed in rhododendron species due to deficient in genomic information. Therefore, it is important to develop more SSR markers. In this study, a total of 8266 SSRs from the transcriptome of R. molle flower were identified. The dinucleotide repeat motifs were the most frequent SSR motifs. The TC/GA/AG/CT motifs were the most frequency motifs, accounting for 92.11% of the total dinucleotide SSRs. Among the di-nucleotide repeat motifs, GA repeats were the most common, which is different from that of Arabidopsis in which AG repeats were the most frequent (Lawson and Zhang 2006). It may due to the introduction of additional repeats during chromosome replication (Eckert et al. 2002). Furthermore, (GA)n serve as regulatory elements that contains a series of overlapped GAG motifs involved in light regulation (Liu et al. 2012). The unigenes obtained from R. molle transcriptome have provided a valuable genetic resource for SSRs mining and applications in gene mapping, cultivar fingerprinting, and selection of desirable genotypes in rhododendron breeding programs.
We conducted Illumina sequencing to profile the transcriptome of R. molle. To the best of our knowledge, this study presents the first next-generation sequencing of R. molle. The resource information would lay an important foundation for future genetic or genomic studies on rhododendron species. Meanwhile, a large number of putative R. molle homologs that are probably involved in carotenoid biosynthesis pathway were identified. We have identified a large amount of SSRs that are ready for marker development.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bruni I, De Mattia F, Labra M, Grassi F, Fluch S, Berenyi M, Ferrari C (2012) Genetic variability of relict Rhododendron ferrugineum L. populations in the Northern Apennines with some inferences for a conservation strategy. Plant Biosyst 146:24–32
Chen L, Li LN, Dai YP, Wang XR, Duan YF, Yang GD (2015) De novo transcriptome analysis of Osmanthus serrulatus Rehd. flowers and leaves by Illumina sequencing. Biochem Syst Ecol 61:531–540
Chen H, Chen X, Tian J, Yang Y, Liu Z, Hao X, Wang L, Wang S, Liang J, Zhang L et al (2016) Development of gene-based SSR markers in rice bean (Vigna umbellata L.) based on transcriptome data. PLoS ONE 11:e0151040
Chosson E, Chaboud A, Chulia AJ, Rayhaud J (1998) Dihydroflavonol glycosides from Rhododendron ferrugineum. Phytochemistry 49:1431–1433
Eckert KA, Mowery A, Hile SE (2002) Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry 41:10490–10498
Gismondi A, Canini A (2013) Microsatellite analysis of Latial Olea europaea L. cultivars. Plant Biosyst 147:686–691
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Joaquín MT, Conesa DA (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Guo LN, Zhao XL, Gao XF (2016) De novo assembly and characterization of leaf transcriptome for the development of EST-SSR markers of the non-model species Indigofera szechuensis. Biochem Syst Ecol 68:36–43
Han Y, Wan HH, Cheng TR, Wang J, Yang WR, Pan HT, Zhang QX (2017) Comparative RNA-seq analysis of transcriptome dynamics during petal development in Rosa chinensis. Sci Rep 7:43382
Hirschberg J (2001) Carotenoid biosynthesis in flowering plants. Curr Opin Plant Biol 4:210–218
Huyen DTT, Ureshino K, Van DT, Miyajima I (2016) Co-pigmentation of anthocyanin-flavonol in the Blotch Area of Rhododendron simsii Planch. flowers. Hortic J 85:232–237
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T et al (2008) Kegg for linking genomes to life and the environment. Nucleic Acids Res 36:D480-D484
Lawson MJ, Zhang LQ (2006) Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol 7:R14
Li X, Lu M, Tang D, Shi Y (2015) Composition of carotenoids and flavonoids in narcissus cultivars and their relationship with flower color. PLoS ONE 10:e0142074
Li J, Lv X, Wang L, Qiu Z, Song X, Lin J, Chen W (2017) Transcriptome analysis reveals the accumulation mechanism of anthocyanins in ‘Zijuan’ tea (Camellia sinensis var. asssamica (Masters) kitamura) leaves. Plant Growth Regul 81:51–61
Liu MY, Qiao GR, Jiang J, Yang HQ, Xie LH, Xie JZ, Zhuo RY (2012) Transcriptome sequencing and de novo analysis for Ma Bamboo (Dendrocalamus latiflorus Munro) using the Illumina platform. PLoS ONE 7:e46766
Luo H, Sun C, Li Y, Wu Q, Song J, Wang D, Jia X, Li R, Chen S (2010) Analysis of expressed sequence tags from the Huperzia serrata leaf for gene discovery in the areas of secondary metabolite biosynthesis and development regulation. Physiol Plant 139:1–12
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12:671–682
Miyajima I, Ureshino K, Kobayashi N, Akabane M (2000) Flower color and pigments of intersubgeneric hybrid between white-flowered evergreen and yellow-flowered deciduous azaleas. J Jpn Soc Hortic Sci 69:280–282
Mizuta D, Ban T, Miyajima I, Nakatsuka A, Kobayashi N (2009) Comparison of flower color with anthocyanin composition patterns in evergreen azalea. Sci Hortic 122:594–602
Nakatsuka A, Mizuta D, Kii Y, Miyajima I, Kobayashi N (2008) Isolation and expression analysis of flavonoid biosynthesis genes in evergreen azalea. Sci Hortic 118:314–320
Ohmiya A (2013) Qualitative and quantitative control of carotenoid accumulation in flower petals. Sci Hortic 163:10–19
Ohmiya A, Kishimoto S, Aida R, Yoshioka S, Sumitomo K (2006) Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in chrysanthemum petals. Plant Physiol 142:1193–1201
Pandurangaiah S, Ravishankar KV, Shivashankar KS, Sadashiva AT, Pillakenchappa K, Narayanan SK (2016) Differential expression of carotenoid biosynthetic pathway genes in two contrasting tomato genotypes for lycopene content. J Biosciences 41:257–264
Rezaei MK, Deokar A, Tar’an B (2016) Identification and expression analysis of candidate genes involved in carotenoid biosynthesis in chickpea seeds. Front Plant Sci 7:1867
Sagawa JM, Stanley LE, LaFountain AM, Frank HA, Liu C, Yuan Y-W (2016) An R2R3-MYB transcription factor regulates carotenoid pigmentation in Mimulus lewisii flowers. New Phytol 209:1049–1057
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29:22–28
Ureshino K, Nakayama M, Miyajima I (2016) Contribution made by the carotenoid cleavage dioxygenase 4 gene to yellow colour fade in azalea petals. Euphytica 207:401–417
Varshney RK, Graner A, Sorrells ME (2005a) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Varshney RK, Sigmund R, Borner A, Korzun V, Stein N, Sorrells ME, Langridge P, Graner A (2005b) Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci 168:195–202
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang P, Ma Y, Ma L, Li Y, Wang S, Li L, Yang R, Wang Q (2016) Development and characterization of EST-SSR markers for Catalpa bungei (Bignoniaceae). Appl Plant Sci 4:1500117
Wang JJ, Wang HB, Ding L, Song AP, Shen F, Jiang JF, Chen SM, Chen FD (2017) Transcriptomic and hormone analyses reveal mechanisms underlying petal elongation in Chrysanthemum morifolium ‘Jinba’. Plant Mol Biol 93:593–606
Xia EH, Zhang HB, Sheng J, Li K, Zhang QJ, Kim C, Zhang Y, Liu Y, Zhu T, Li W et al (2017) The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol Plant 10:866–877
Xiao Z, Sun X, Liu X, Li C, He L, Chen S, Su J (2016) Selection of reliable reference genes for gene expression studies on Rhododendron molle G. Don. Front Plant Sci 7:1547
Yan J, Cai ZX, Shen ZJ, Zhang BB, Ma RJ, Yu ML (2015) Extraction and analytical methods of carotenoids in fruit of yellow flesh peach. J Fruit Sci 32:1267–1274
Yang LE, Huang XQ, Lu QQ, Zhu JY, Lu S (2016) Cloning and characterization of the geranylgeranyl diphosphate synthase (GGPS) responsible for carotenoid biosynthesis in Pyropia umbilicalis. J Appl Phycol 28:671–678
Yuan H, Zhang J, Nageswaran D, Li L (2015) Carotenoid metabolism and regulation in horticultural crops. Hortic Res 2:15036
Zhang B, Liu C, Wang Y, Yao X, Wang F, Wu J, King GJ, Liu K (2015) Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol 206:1513–1526
Zhang YZ, Xu SZ, Cheng YW, Ya HY, Han JM (2016) Transcriptome analysis and anthocyanin-related genes in red leaf lettuce. Genet Mol Res 15:gmr.15017023
Acknowledgements
This work was supported by the National Science and Technology Program (2013BAD01B070403), National Natural Science Foundation of China (31600570), Jiangsu Natural Science Foundation (BK20150548), Jiangsu Forestry Sciences Innovation Fund (lykj[2017]48), and Jiangsu Agricultural Sciences Innovation Fund (CX(16)1005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Zheng Xiao declares that he does not have conflict of interest. Jiale Su declares that he does not have conflict of interest. Xiaobo Sun declares that she does not have conflict of interest. Chang Li declares that she does not have conflict of interest. Lisi He declares that she does not have conflict of interest. Shangping Cheng declares that he does not have conflict of interest. Xiaoqing Liu declares that she does not have conflict of interest.
Research involving human and animal participants
This article does not contain any studies with human subjects or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiao, Z., Su, J., Sun, X. et al. De novo transcriptome analysis of Rhododendron molle G. Don flowers by Illumina sequencing. Genes Genom 40, 591–601 (2018). https://doi.org/10.1007/s13258-018-0662-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0662-8