
Abstract
Two catabolite repressor genes (MIG1 and MIG2) were previously identified in Pichia pastoris, and the derepression of alcohol oxidase (AOX) expression was realized in Δmig1 or Δmig1Δmig2 mutants grown in glycerol, but not in glucose. In this study, genome-wide RNA-seq analysis of Δmig1Δmig2 and the wild-type strain grown in glycerol revealed that the expression of numerous genes was greatly altered. Nearly 7% (357 genes) of approximately 5276 genes annotated in P. pastoris were significantly upregulated, with at least a two-fold differential expression in Δmig1Δmig2; the genes were mainly related to cell metabolism. Approximately 23% (1197 genes) were significantly downregulated; these were mainly correlated with the physiological characteristics of the cell. The methanol catabolism and peroxisome biogenesis pathways were remarkably enhanced, and the genes AOX1 and AOX2 were upregulated higher than 30-fold, which was consistent with the experimental results of AOX expression. The Mig proteins had a slight effect on autophagy when cells were grown in glycerol. The expression analysis of transcription factors showed that deletion of MIG1 and MIG2 significantly upregulated the binding of an essential transcription activator, Mit1p, with the AOX1 promoter, which suggested that Mig proteins might regulate the AOX1 promoter through the regulation of Mit1p. This work provides a reference for the further exploration of the methanol induction and catabolite repression mechanisms of AOX expression in methylotrophic yeasts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The methylotrophic yeast Pichia pastoris has been developed as an effective expression system for various heterologous proteins (Cereghino and Cregg 2000; Gasser et al. 2013). This organism is especially attractive as it possesses a strong and tightly regulated methanol-inducible promoter of the alcohol oxidase 1 gene (AOX1) (P AOX1 ) and grows to reach high cell densities (Li et al. 2007; Vogl and Glieder 2013). P AOX1 is induced only in response to methanol, but repressed in alternative carbon sources, such as glucose or glycerol (Hartner and Glieder 2006).
In methylotrophic yeasts, a diauxic cell usually appears in a mixture of glycerol/methanol or glucose/methanol without the repression of the methanol- utilizing enzymes, such as alcohol oxidase (AOX), catalase (CAT), and dihydroxyacetone synthase (DAS) (Egli et al. 1980; Inan and Meagher 2001). As previously reported, AOX1 expression in P. pastoris is almost undetectable in glycerol (Hartner and Glieder 2006; Inan and Meagher 2001), but exhibits ~ 3–30 and 60–80% derepression, respectively, in Candida boidinii and Hansenula polymorpha (Hartner and Glieder 2006; Sasano et al. 2010). Although the phenomenon of catabolite repression in yeasts has been known for a long time, there are only a few reports that describe the regulatory mechanism of the repression of AOX synthesis in methylotrophic yeasts (Leão-Helder et al. 2004; Stasyk et al. 2008; Zhang et al. 2010). As previously reported, some aspects of catabolite repression are mediated by a Cys2His2 zinc-finger transcription factor, Mig1p/CreA, which specifically binds to DNA with a GC-rich consensus sequence and a flanking AT sequence, conserved in most eukaryotic species (Kulmburg et al. 1993; Mathieu and Felenbok 1994; Nehlin and Ronne 1990; Nehlin et al. 1991). In Saccharomyces cerevisiae, Mig1p functions to repress the transcription of genes under conditions of glucose sufficiency, such as those genes encoding enzymes for the utilization of the sugars galactose, sucrose, or maltose (Carlson 1999; Schüller 2003). In the methanol-utilizing yeast H. polymorpha, the researchers developed conditions mimicking the molecular pathway of catabolite repression of the conventional yeast S. cerevisiae (the so-called Snf1-Mig1 pathway) (Carlson 1999; Gancedo 1998) to identify and study the role of transcriptional repressors HpMig1p and HpMig2p in catabolite repression. The repression pathway for AOX in this yeast partially relies on the two Mig1p homologs, irrespective of the carbon source (glucose, sucrose, or ethanol) (Stasyk et al. 2007). P. pastoris and H. polymorpha, which are regarded to be belonging to the same taxonomic position, were identified to have methanol-assimilating capabilities (Hazeu et al. 1972). The methanol utilization pathway and the regulation of alcohol oxidase genes are similar in both strains (Hartner and Glieder 2006; Yurimoto et al. 2011). Thus, the regulation of catabolite repression of P AOX1 in P. pastoris may also be related to Mig1p-mediated transcriptional repression. We therefore cloned two Mig1p homologs (designated Mig1p and Mig2p) in P. pastoris (Wang et al. 2017). Gene knock-out analysis showed that the double-deletion of MIG1 and MIG2 allowed the expression of alcohol oxidase under glycerol (Wang et al. 2017), which was similar to findings in H. polymorpha (Stasyk et al. 2007).
However, it is unknown how these Mig proteins affect global cell metabolism, especially methanol catabolism and the closely related peroxisome biosynthesis. The next-generation sequencing technology for RNA (RNA-seq) is a useful and cost-efficient tool for transcriptome analysis (Marguerat and Bähler 2010), which offers better dynamic range, detects very subtle changes in gene expression, characterizes the alternative splicing of mRNAs, and detects novel transcripts (Ozsolak and Milos 2010; Wilhelm and Landry 2009). Thus, RNA-seq based transcriptome analysis was used in this study to plot the genetic and metabolic variations of the Δmig1Δmig2 mutant grown in glycerol with P AOX1 derepression in comparison with the wild type strain, which may help us clarify the regulatory roles of Mig proteins.
Materials and methods
Strains, growth conditions and microbial techniques
The strains used in this study were listed in Table S1. The cells were grown on standard liquid or solid media at 30 °C as previously described (Zhang et al. 2010). The concentration of methanol in medium was 0.5% (v/v) while that of any other carbon source was 1% (w/v) unless indicated otherwise. Cell density was determined by measuring absorbance at 600 nm. Construction of strains of Δmig1, Δmig2 and Δmig1Δmig2 were described previously (Wang et al. 2017).
Analysis of cell growth and AOX activity
Cells of each strain were preincubated overnight in YNB medium supplemented with glucose (1%, v/v) and transferred into fresh YNB medium supplemented with various carbon sources (1% glucose, 1% glycerol, 0.5% methanol, 1% glycerol + 0.5% methanol) at an initial OD600 of 0.03. Preparation of crude cell extracts and assays of AOX activity were carried out as described previously (Zhang et al. 2010). The AOX activity was detected since 12 h culture of cells on various carbon sources (Zhang et al. 2010) to collect enough cells for analysis.
Strains and culture conditions for RNA-seq
The following P. pastoris strains were used for RNA-seq analysis: wild type GS115 and Δmig1Δmig2 mutant (Wang et al. 2017). P. pastoris cells were grown in yeast nitrogen base (YNB) with adequate auxotrophic requirements containing 1% glucose or 1% glycerol as carbon source in batch cultures. For transcriptome analysis, cells were pre-cultured in glucose at 30 °C prior to a shift to fresh media containing glycerol as sole carbon source. Four independent glucose-grown cultures (two parallel samples for each strain) were used to inoculate into fresh medium containing glycerol. As previously reported, transcription of genes involved in methanol catabolism varied significantly even after culture in glucose or glycerol for only 2 h. Thus the glycerol cultures (initial OD600 of 1.0) grown for 4 h, which led to comparative cell density to cells grown for 12 h in the cell growth and AOX activity analytic experiments, were used for RNA isolation.
RNA preparation
Total RNA was extracted using Ribopure Yeast kit (Ambion, Austin, TX) and treated for 30 min at 37 °C with RNase free DNase I (Ambion) for removing residual genomic DNA. The quality and quantity of the purified RNA were determined with a Nanodrop ND-1000 spectrophotometer (LabTech, USA) by measuring absorbance at 260 nm/280 nm (A260/A280). RNA integrity was further verified by agarose gel electrophoresis.
Preparation of cDNA library for RNA-seq
The cDNA libraries were prepared according to the manufacturer’s instructions (Illumina). Poly (A) mRNA was isolated from the total RNA samples with oligo (dT) magnetic beads (Invitrogen). To avoid priming bias when synthesizing cDNA, the mRNA was firstly fragmented before cDNA synthesis. The purified mRNA was fragmented into small pieces by the RNA fragmentation kit (Ambion) and applied as template for first-strand cDNA synthesis using random hexamer primers and reverse transcriptase (Invitrogen). The second-strand cDNA was synthesized using RNase H (Invitrogen) and DNA polymerase I (New England Biolabs, USA). The Illumina Genomic DNA Sample Prep kit (Illumina, USA) was used to generate 95-bp paired-end (PE) cDNA libraries. The libraries were loaded onto flow cell channels for sequencing on the Illumina GA IIx system. A total of four paired-end cDNA libraries of P. pastoris cells were constructed for each of the test groups. Duplicate technical replicates were performed for each group.
Mapping reads to reference genome and annotated genes
The 95-bp raw PE reads (400 bp insert size), generated by the Illumina GA IIx system, were firstly processed by the FASTX-Toolkit to remove the reads containing sequencing adapters and reads of low quality (phred quality < 5) and then aligned the remaining reads using the EMBL P. pastoris GS115 coding sequence database as reference by the Burrows–Wheeler Aligner’s Smith–Waterman Alignment (BWA) program (Li and Durbin 2010).
Data deposit
The original sequencing is deposited in SRA (Short Read Archive) with the accession number SRP094666 in the National Center for Biotechnology Information database (NCBI, http://www.ncbi.nlm.nih.gov).
Normalized expression level of gene by RNA-seq
The gene expression level by RNA-seq was normalized by the number of reads per kilobase of exon region per million mapped reads (RPKM) using the formula described by Mortazavi et al. (2008). The cut-off value for determining gene transcriptional activity was determined based on a 95% confidence interval for all RPKM values of each gene. Two biological replicates of each sample were used for differential gene expression analysis between the Δmig1Δmig2 mutant and WT strain. A 0.81–0.88 Pearson correlation coefficient was obtained for biological replicates.
Differential expression analysis
The R package DEGseq (Wang et al. 2010) was used to identify differentially expressed genes in different strains with a ratio of RPKM in Δmig1Δmig2 to WT, higher than 2.0 or lower than 0.5 (log2 (ratio) ≥ 1 or ≤ − 1) and p value < 0.05 [cut-off at 5% false discovery rate (FDR)]. FDR (Benjamini and Hochberg 1995) was performed using the default parameters to obtain an adjusted p-value between the Δmig1Δmig2 group versus the WT group. Then the resulting expression intensity values were analyzed by the MA plot-based method, as described by Wang et al. (2010).
Gene ontology and KEGG pathway analysis
The ORF-encoded proteins were categorized according to the P. pastoris genome gene ontology (GO) annotations from NCBI (http://www.ncbi.nlm.nih.gov/), UniProt (http://www.uniprot.org/) and the GO (http://www.geneontology.org/). The “elim Fisher” algorithm (Alexa et al. 2006) was used for GO enrichment test because it can iteratively remove the genes mapped to significant GO terms from more general (higher level) GO terms so that the significant GO terms will not be overshadowed by the general GO terms. GO categories with a p value < 0.01 were reported.
Pathway analysis was used to find out the significant pathway of the differential genes. Pathway annotations of RNA-seq genes were downloaded from KEGG (Kyoto Encyclopedia of Genes and Genomes) (http://www.genome.jp/kegg/). A Fisher exact test was used to find the significant enrichment pathway. The resulting p values were adjusted using the FDR algorithm (Benjamini and Hochberg 1995). Pathway categories with a FDR < 0.05 were reported. Enrichment provides a measure of the significance of the function: as the enrichment increases, the corresponding function becomes specific, which helps us to find those more significant pathways in the experiment. The enrichment was given by: enrichment = \({{\left( {{{{n_g}} \mathord{\left/ {\vphantom {{{n_g}} {{n_a}}}} \right. \kern-0pt} {{n_a}}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{{n_g}} \mathord{\left/ {\vphantom {{{n_g}} {{n_a}}}} \right. \kern-0pt} {{n_a}}}} \right)} {\left( {{{{N_g}} \mathord{\left/ {\vphantom {{{N_g}} {{N_a}}}} \right. \kern-0pt} {{N_a}}}} \right)}}} \right. \kern-0pt} {\left( {{{{N_g}} \mathord{\left/ {\vphantom {{{N_g}} {{N_a}}}} \right. \kern-0pt} {{N_a}}}} \right)}}\) where n g is the number of differential genes within the particular pathway, n a is the total number of genes within the same pathway, N g is the number of differential genes which have at least one pathway annotation, and N a is the number of genes which have at least one pathway annotation in the entire RNA-seq.
Validation of RNA-seq analysis by quantitative real-time PCR
Real-time quantitative PCR (qPCR) was performed to validate gene transcription levels determined by RNA-seq. The P. pastoris GS115 samples for qPCR were as same as those for Illumina sequencing. Total RNA (1 μg) was reversely transcribed into cDNA using PrimeScript™ RT-PCR Kit (TaKaRa). Each 20 μl reaction solution contained primers (optimal concentration), SYBR Premix Ex Taq (TaKaRa, Dalian, China) and cDNA templates (1/100 dilution), and was performed on a 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). The primers for qPCR (Table S2) were designed using Primer Express software (Applied Biosystems, Foster City, CA), with predicted products in the 100- to 300-bp size range. The cycle threshold values (CT) were determined and the relative fold differences were calculated by the 2−ΔΔCT method (Schmittgen and Livak 2008) using ACT1 as the endogenous reference genes (Vandesompele et al. 2002). Samples were run in triplicate in a 96-well plate and each experiment was repeated three times. Two experiments of independently cultivated yeast were performed to confirm the reproducibility of the results.
Results
Regulation of cell growth and catabolite repression of AOX expression by P. pastoris Mig1p, Mig2p
In our previous study (Wang et al. 2017), two genes coding for transcription repressors, MIG1 (PAS_chr4_0334) and MIG2 (PAS_chr1-4_0526), were identified via the P. pastoris genome database (http://bioinformatics.psb.ugent.be/webtools/bogas/overview/Picpa) (De Schutter et al. 2009). To determine the functions of Mig1p and Mig2p, the Δmig1, Δmig2, and Δmig1Δmig2 strains were constructed (Wang et al. 2017), and derepression of alcohol oxidase (AOX) expression was realized in Δmig1 or Δmig1Δmig2 grown in glycerol, but not in glucose. However, as this previous work focused on the metabolic engineering of P. pastoris for the development of a methanol-free expression system, our main interest was whether the derepression of AOX expression was realized; consequently, the bioinformatic analysis of Mig proteins and the regulation of cell growth and derepression of AOX expression by Mig proteins were not systematically analyzed.
Phylogenetic analysis revealed that Mig1p and Mig2p are members of the subgroup of transcriptional repressors that contain selected homologs of Mig proteins and the fungal CreA protein. Interestingly, the distance of the branch between P. pastoris Mig1p and H. polymorpha Mig1p (HpMig1p) (Stasyk et al. 2007) is short, as is that between PpMig2p and H. polymorpha Mig2p (HpMig2p) (Stasyk et al. 2007) (Fig. S1A), which implied that the two putative transcriptional repressors may play similar roles in catabolite repression in P. pastoris as HpMig1p and HpMig2p in H. polymorpha. The translated protein sequences of P. pastoris Mig1p and Mig2p have 444 and 454 amino acid residues, respectively. The amino acid sequences contain conserved Mig1p/CreA/CRE1 analogs from different microorganisms, but also have some distinctive features. Both Mig proteins contain two Cys2His2 zinc-finger motifs, which harbor a putative phosphorylation sequence for the cAMP-dependent protein kinase located in the second zinc-finger region (Fig. S1B). P. pastoris Mig2p contains a stretch rich in glutamine, near amino acids 159 and 186 (Fig. S1C), which appears in several proteins that are presumed to regulate gene expression (Schultz and Carlson 1987), but similar regions cannot be found in Mig1p. Moreover, an additional conserved region was found in yeast Mig proteins (Fig. S1D): the so-called C-terminal “effector domain” (Ostling et al. 1996) existed in Mig1p and Mig2p, although the protein sequence differed in Mig1p and Mig2p (LPSLSSL and LPPIRSL, respectively) (Fig. S1E).
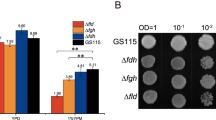
The lack of MIG1 strongly damaged cell growth, especially in the presence of glucose and glycerol, whereas the deficiency of MIG2 resulted in only mild damage (Fig. 1). The double deletion of MIG1 and MIG2 caused more severe effects, with severe inhibition of cell growth in various carbon sources (Fig. 1). To gain further insight into the repressive effects of Mig1p and Mig2p triggered by rich carbon sources, we assayed the AOX activity in the mutants. A glucose concentration of 1% highly repressed AOX expression during the whole culture process, regardless of whether MIG1 or MIG2 genes were deleted (Fig. 1a). However, Δmig1 resulted in modest AOX expression during the late phase; and interestingly, Δmig1Δmig2 showed a strong derepression of AOX expression in 1% glycerol compared with the WT, in which AOX expression was almost completely repressed (Fig. 1b). Catabolite repression by glycerol was further relieved when the cells were cultivated in 1% glycerol supplemented with 0.5% methanol (glycerol + methanol) (Fig. 1c, d). For both the Δmig2 and Δmig1Δmig2 mutants grown in glycerol and glycerol + methanol, the AOX activity reached a high level and then decreased gradually to almost zero before 40 h (Fig. 1b, d). This was different to strains cultured with methanol as the sole carbon source, in which the AOX activity remained at a high level for up to 40 h, despite the sharp decrease later caused by methanol depletion.

Relative expression levels of P. pastoris (wild type and mutants) alcohol oxidase during growth on various carbon sources: a 1% glucose; b 1% glycerol; c 0.5% methanol; d 1% glycerol + 0.5% methanol
Summary of RNA-seq data sets
To explore how Mig proteins affected the Δmig1Δmig2 mutant, cultured in glycerol showing P AOX1 derepression, and its related physiological process in comparison with their effect on the wild type strain, transcriptome analysis based on RNA-seq was conducted on the wild type (W) and Δmig1Δmig2 double-deletion mutant (S) grown in glycerol. The ORFs in the RNA-seq dataset for differential expression analysis involved 5277 of the mapped or unmapped ORFs in the reference genome. Each group contained two biological replicates, which showed good correlation (Fig. S2A), and the data were combined for further analysis. In total, 26,247,210 reads were obtained and a close agreement between the biological replicates was confirmed by the Pearson correlation coefficients of 0.952 (W) and 0.962 (S) (Table S3).
Transcriptional comparative analysis reveals differentially expressed genes between Δmig1Δmig2 and wild type
To assess differences more accurately between transcriptome data in Δmig1Δmig2 versus wild type, the data were analyzed by the DEGseq package in R software (Wang et al. 2010). A two-fold or greater change in expression and p value < 0.05 (cut-off at 5% FDR) was determined as significant. An MA-plot of differentially expressed genes is shown in Fig. S2B. A comparative analysis of the RNA-seq data revealed that 1554 genes were differentially expressed and 357 genes were upregulated by at least two-fold when AOX1 and AOX2 were increased by more than 30-fold. GO analysis revealed that the genes upregulated in the double-mutant strain were mainly involved in cell metabolism (Fig. 2a), whereas the downregulated genes appeared to be mainly associated with cell physiology processes (Fig. 2b). In addition, KEGG metabolic pathway analysis indicated that the genes upregulated in double-mutant strain were located specifically in the pathways of the metabolic processes: biosynthesis of secondary metabolites; glycolysis and gluconeogenesis; pyruvate metabolism; glyoxylate and dicarboxylate metabolism; and the citrate cycle. Conversely, the downregulated genes were mainly associated with ribosomes, biosynthesis of secondary metabolites, and oxidative phosphorylation (Fig. 3). Generally, KEGG pathway analysis showed similar results to the GO analysis; that is, substance metabolism in the cell was enhanced, whereas DNA and protein synthesis and cell division were reduced by the deletion of MIG1 and MIG2 grown in glycerol.

Significantly upregulated (a) and downregulated (b) genes and their assigned gene ontology (GO) categories in P. pastoris

Statistically significant KEGG classifications of differentially expressed genes in P. pastoris
Enhancement of methanol catabolism and peroxisome biogenesis pathways in Δmig1Δmig2 mutant
In Fig. 4, an overview of the methanol metabolism (Van Der Klei et al. 2006), including a ratio of RPKM in Δmig1Δmig2 mutant to that in WT, was presented. In peroxisomes, methanol was oxidized to formaldehyde and hydrogen peroxide by AOX, which was 32.6 times upregulated at the transcriptional level, which also supported the AOX expression results shown in Fig. 1b. Hydrogen peroxide was detoxified by catalase (CAT) to water and oxygen (1.5-fold upregulation). Formaldehyde can be further metabolized via two different routes: dissimilation via formaldehyde dehydrogenase (FLD1, 1.2-fold upregulation), S-formyl glutathione hydrolase (FGH, 0.9-fold upregulation), and formate dehydrogenase (FMD, 2.4-fold upregulation) to CO2, generating NADH and CO2, or assimilation via the peroxisome-borne enzyme dihydroxyacetone synthase (DHAS, 6.2-fold upregulation) to generate cell constituents. DHAS is part of the xylulose-5-phosphate cycle and catalyzes the formation of two C3-molecules (dihydroxyacetone and glyceraldehyde-3-phosphate) from one C1 (formaldehyde) and one C5 (xylulose-5-phosphate) compound. Thus, methanol catabolism was highly upregulated by the deletion of MIG1 and MIG2 in P. pastoris grown in glycerol.

Significantly differentially expressed genes in the methanol utilization pathway in Pichia pastoris. The schematic diagram was adapted from Van Der Klei et al. (2006). AOX alcohol oxidase, FLD formaldehyde dehydrogenase, FGH S-formylglutathione hydrolase, FDH formate dehydrogenase, CAT catalase, DAS dihydroxyacetone synthase, DAK dihydroxyacetone kinase, TPI triosephosphate isomerase, FBA fructose-1,6-bisphosphate aldolase, FBP fructose-1,6-bisphosphatase, DHA dihydroxyacetone, GAP glyceraldehyde-3-phosphate, DHAP dihydroxyacetone phosphate, F1,6BP fructose-1,6-bisphosphate, F6P fructose-6-phosphate, Pi phosphate, Xu5P xylulose-5-phosphate, GSH glutathione. Red numbers indicate the fold- change of the upregulated genes in the Δmig1Δmig2 strain compared with the WT strain, and blue numbers indicate the fold- change of downregulated genes in the Δmig1Δmig2 strain compared with the WT strain. (Color figure online)
AOX synthesis is closely related to peroxisome biogenesis (Distel et al. 1988; Ozimek et al. 2005). PEX genes control the development and function of the peroxisomes (Sibirny 2016). Clear upregulations were observed for PEX1 (2.6-fold), PEX5 (2.6-fold), PEX6 (3.2-fold), PEX14 (2.3-fold), all of which were correlated with the import of peroxisomal matrix enzymes (AOX, DHAS, and CAT), as shown by peroxisome biogenesis in KEGG (Fig. 5). Additionally, a peroxisome membrane protein- encoded gene, PMP47B, was also highly upregulated (2.75-fold). Moreover, most of the PEX genes were moderately increased in Δmig1Δmig2 grown in glycerol (Table S4).

Significantly differentially expressed genes related to the peroxisome biosynthesis pathway in Pichia pastoris. The pathway is referred to that of Saccharomyces cerevisiae from KEGG pathway database. Red numbers indicate the fold change of upregulated genes in Δmig1Δmig2 strain compared with the WT strain and blue numbers indicate the fold change of downregulated genes in the Δmig1Δmig2 strain compared with the WT strain. (Color figure online)
Effects of MIG1 and MIG2 double deletion on autophagy
The adaptation of P. pastoris cells to methanol or other carbon sources requires a major rearrangement of the cellular architecture. Several ATG (autophagy related gene) genes (ATG1, ATG5, ATG12, ATG16, ATG27, and ATG28) were downregulated by more than two-fold, but only one gene (ATG26) was upregulated by more than two-fold (Table S5), which indicated that autophagy in the Δmig1Δmig2 mutant was ameliorated to a certain extent and proved that Mig proteins may also act as a regulator of autophagy in P. pastoris. Interestingly, other pexophagy related genes, ATG30 (Farré et al. 2008), ATG25 (Monastryska et al. 2005), and ATG9 (Chang et al. 2005), were not clearly different between the wild- type strain and the Δmig1Δmig2 mutant. Additionally, ATG8, which is related to various selective and non-selective macroautophagic processes, and ATG11, which is involved in selective macroautophagy (Carlson 1999; Carmona et al. 2002), also showed no clear differences in either strain grown in glycerol. Overall, Mig1p and Mig2p only slightly affected autophagy when the cells were grown in glycerol.
Effects of MIG1 and MIG2 double deletion on expression of transcription factors
As transcription repressors, Mig1p and Mig2p might function as transcription factors for the direct regulation of P AOX1 , or the indirect regulation of the expression of transcription factors of P AOX1 . Therefore, we analyzed the variation in expression of the transcription factors in the KEGG database (http://www.genome.jp/kegg-bin/get_htext#A1) in Δmig1Δmig2. As shown in Table 1, two genes (2.17-fold for PAS_chr2-1_0757; 2.38-fold for PAS_chr3_0836) were significantly upregulated, whereas three genes (2.75-fold for PAS_chr2-1_0206; 2.84-fold for PAS_chr1-4_0308; 2.62-fold for PAS_chr1-4_0183) were significantly downregulated. Among them, the PAS_chr3_0836 gene encodes a critical transcription activator, Mit1p, of P AOX1 in P. pastoris, which has been previously verified by our group (Wang et al. 2016). In addition, the deletions of MIG1 and MIG2 did not significantly influence the two other identified transcription factors of P AOX1 , Mxr1 (Lin-Cereghino et al. 2006) and Prm1 (Sahu et al. 2014). The possible transcription factor binding sites of P AOX1 and P MIT1 were then analyzed by the online software AliBaba2.1 (http://gene-regulation.com/pub/programs/alibaba2/index.html?), which works by the construction of the matrices on the fly from TRANSFAC 4.0 sites (http://gene-regulation.com/pub/databases.html#transfac). The scanning results indicated that P MIT1 holds two Mig1-binding motifs (5′-ATCTTCCCCGCC-3′; 5′-CCTCCCCACA-3′), which contain conserved regions with the reported Mig1/CreA binding sites in S. cerevisiae and Aspergillus nidulans (Kulmburg et al. 1993; Mathieu and Felenbok 1994; Nehlin and Ronne 1990; Nehlin et al. 1991). However, P AOX1 showed no Mig1-binding motifs by the online promoter scanning. Thus, Mig proteins probably repress P AOX1 through the negative regulation of Mit1p expression. Moreover, by BLAST searching, we found that the PAS_chr3_0757 gene encodes a protein homolog of the transcription activator, Cat8 in S. cerevisiae, which encodes a transcription activator for the expression of a glucose-repressible alcohol dehydrogenase (Hedges et al. 1995; Walther and Schüller 2001). In S. cerevisiae, Mig1p also suppressed Cat8 (Hedges et al. 1995). However, the deletion of this gene did not affect AOX expression in P. pastoris (data not shown). As for the three downregulated genes, their specific relationship with AOX expression could not be identified with our present knowledge. Thus, Mig proteins may have a broad function in gene regulation related to carbon catabolism and other biological processes in P. pastoris.
Validation of unigenes and gene expression profiling
In order to experimentally validate the reliability of gene expression obtained by RNA-seq data, seven genes were selected and their transcription levels were confirmed through the measurement of the relative mRNA levels by using quantitative RT-PCR with the primers listed in Table S2. Our results, shown in Fig. 6, indicated that the data from qRT-PCR were predominantly consistent with those of RNA-seq, which confirmed the validation of the RNA-seq data and certified the reliability of the RNA-seq data. The differences in the relative fold- change in transcription between qRT-PCR and RNA-seq were mainly ascribed to the biases of the different primers.

Quantitative RT-PCR validations for RNA-seq data in Pichia pastoris. A total of seven genes were selected for the quantitative qRT-PCR experiments. AOX1 (PAS_chr4_0821), AOX2 (PAS_chr4_0152), DAS1 (PAS_chr3_0832), and DAS2 (PAS_chr3_0834) were used for the methanol metabolism pathway; MIT1 (PAS_chr3_0836), PRM1 (PAS_chr4_0203), and MXR1 (PAS_chr4_0487) were transcription factors. One-way analysis of variance (Origin 7.0, OriginLab Corporation, USA) was used to determine the significant difference of gene transcription in quantitative RT-PCR experiments between the wild type and Δmig1Δmig2 strains. The significance levels were at *p < 0.05, **p < 0.01, and ++ p > 0.05
Discussion
The methylotrophic yeast P. pastoris has been widely used for various heterologous proteins because of its powerful protein expression ability driven by P AOX1 . However, the transcription regulation networks of P AOX1 regarding methanol induction and catabolite repression have not been fully elucidated.
As a transcription factor, Mig1p has been identified to participate in catabolite repression in S. cerevisiae (Carlson 1999; Schüller 2003). Moreover, in the methanol-utilizing yeast H. polymorpha, the repression pathway for AOX is only partially reliant on the two Mig1p homologs, irrespective of the carbon source (glucose, sucrose, or ethanol) without glycerol (Stasyk et al. 2007). P. pastoris and H. polymorpha were identified to have similar methanol-assimilating capabilities and regulation mechanisms of the respective alcohol oxidase genes, despite the strict repression of alcohol oxidase genes in P. pastoris (Egli et al. 1980; Hazeu et al. 1972). Therefore, the roles of MIG1 and MIG2 were explored with regard to its regulation on P AOX1 in P. pastoris.
As glycerol is commonly used in P. pastoris fermentation, the roles of MIG1 and MIG2 in the repression of AOX expression in glycerol were considered in the presence of glucose, which was different from the study conducted in H. polymorpha (Stasyk et al. 2007). The double deletion of MIG1 and MIG2 damaged cell growth in glucose and glycerol (Fig. 1), which was also in accordance with the results in H. polymorpha (Stasyk et al. 2007). This might be attributable to the downregulation of the cellular physiological processes reflected in the GO and KEGG analysis results (Figs. 2, 3). Conversely, although the double deletion of MIG1 and MIG2 caused the derepression of AOX expression in glucose in H. polymorpha, it was strictly repressed in P. pastoris, which indicated that catabolite repression was stricter in P. pastoris. However, AOX expression was largely derepressed in Δmig1Δmig2 mutants grown in glycerol.
To completely comprehend the global metabolic changes in Δmig1Δmig2, RNA-seq was applied to generate two high-resolution transcriptional profiles of the WT and Δmig1Δmig2 strains grown with glycerol as the sole carbon source. This provided information on the induction and repression of metabolic genes, as well as non-metabolic genes. Consistent with the AOX activity detected in glycerol medium (Fig. 1b), the methanol catabolism pathway was markedly strengthened, with the AOX1 level increased by more than 30 times (Fig. 4). The results illustrated that Mig proteins highly repressed P AOX1 . Two possible modes may be involved in this process: either by Mig proteins directly bound to P AOX1 ; or the repressed expression of positive transcription factors of P AOX1 or the activated expression of the negative transcription factors of P AOX1 . As the expression of several enzymes other than AOX, i.e., dihydroxyacetone synthase (DAS), formate dehydrogenase (FDH), dihydroxyacetone kinase (DAK), fructose-1,6-bisphosphate aldolase (FBA), were significantly and synchronously upregulated, Mig proteins were more likely to act as an upstream global regulator to control the expression of transcription factors of P AOX1 and the promoters of other upregulated genes in the methanol catabolism pathway. To investigate the possibility that Mig proteins regulated the expression of the transcription factors of P AOX1 , the variations in the expression of the transcription factors between WT and Δmig1Δmig2 were analyzed (Table 1). Two significantly upregulated genes and three downregulated genes coded for the transcription factors were identified. As previously reported, Mit1p was a critical transcription activator of P AOX1 (Wang et al. 2016). Unexpectedly, Mit1p expression was significantly increased in the Δmig1Δmig2 mutant. For another upregulated gene (PAS_chr3_0757), the deletion did not derepress AOX expression (data not shown); thus, it was excluded as a regulator of P AOX1 . Moreover, as for the three downregulated genes, we could not identify a specific relationship between them and AOX expression based on our current knowledge. In S. cerevisiae, Mig1p was verified to bind to the promoter of GAL4, which codes for the transcription activator Gal4 of the GAL genes (Nehlin et al. 1991). Moreover, the online analysis of the transcription factor binding sites predicted that P MIT1 contained two Mig1-binding motifs, similar to the Mig1/CreA binding sites in S. cerevisiae and A. nidulans (Kulmburg et al. 1993; Mathieu and Felenbok 1994; Nehlin and Ronne 1990; Nehlin et al. 1991). Therefore, it could be inferred that Mig1p and Mig2p probably act as transcription repressors of genes, whose transcription was activated by Mit1p. This in turn leads to repression of AOX expression. However, this speculation requires further experiments for verification.
In addition to the enhanced methanol catabolism pathway, the peroxisome biogenesis pathway was also strengthened. As methanol was not used solely in the glycerol medium and AOX was exactly a peroxisomal enzyme in P. pastoris and other methylotrophic yeast it was likely that peroxisome biogenesis was regulated by AOX expression, but not through the direct methanol induction process; however, this requires further study for clarification. In addition, the autophagy in Δmig1Δmig2 appeared to be relieved to some extent, which indicated that Mig proteins might also function in the regulation of autophagy in glycerol in P. pastoris. However, pexophagy was apparently unaffected as the related genes ATG30, ATG25, ATG8, and ATG11 showed no clear changes in different strains grown in glycerol.
Generally, our data show comprehensive information on the role of P. pastoris MIG1 and MIG2 in the derepression of P AOX1 and its related biological processes in P. pastoris. This study provides a useful reference for the further exploration of the methanol induction and catabolite repression mechanisms of P AOX1 , and the future specific and systematic experimental studies of the regulatory mechanisms proposed in this work will allow us to further clarify their roles.
References
Alexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22:1600–1607
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B (Methodological) 57:289–300
Carlson M (1999) Glucose repression in yeast. Curr Opin Microbiol 2:202–207
Carmona TA, Barrado P, Jiménez A, Fernández Lobato M (2002) Molecular and functional analysis of a MIG1 homologue from the yeast Schwanniomyces occidentalis. Yeast 19:459–465
Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66
Chang T, Schroder LA, Thomson JM, Klocman AS, Tomasini AJ, Strømhaug PE, Dunn WA Jr (2005) Ppatg9 encodes a novel membrane protein that traffics to vacuolar membranes, which sequester peroxisomes during pexophagy in Pichia pastoris. Mol Biol Cell 16:4941–4953
De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouzé P, Van de Peer Y, Callewaert N (2009) Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol 27:561–566
Distel B, Van Der Ley I, Veenhuis M, Tabak HF (1988) Alcohol oxidase expressed under nonmethylotrophic conditions is imported, assembled, and enzymatically active in peroxisomes of Hansenula polymorpha. J Cell Biol 107:1669–1675
Egli T, Dijken JP, Veenhuis M, Harder W, Fiechter A (1980) Methanol metabolism in yeasts: regulation of the synthesis of catabolic enzymes. Arch Microbiol 124:115–121
Farré JC, Manjithaya R, Mathewson RD, Subramani S (2008) Ppatg30 tags peroxisomes for turnover by selective autophagy. Dev Cell 14:365–376
Gancedo JM (1998) Yeast carbon catabolite repression. Microbiol Mol Biol R 62:334–361
Gasser B, Prielhofer R, Marx H, Maurer M, Nocon J, Steiger M, Puxbaum V, Sauer M, Mattanovich D (2013) Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol 8:191–208
Hartner FS, Glieder A (2006) Regulation of methanol utilisation pathway genes in yeasts. Microb Cell Fact 5:39
Hazeu W, Bruyn JC, Bos P (1972) Methanol assimilation by yeasts. Arch Microbiol 87:185–188
Hedges D, Proft M, Entian KD (1995) CAT8, a new zinc cluster-encoding gene necessary for derepression of gluconeogenic enzymes in the yeast Saccharomyces cerevisiae. Mol Cell Biol 15:1915–1922
Inan M, Meagher MM (2001) Non-repressing carbon sources for alcohol oxidase (AOX1) promoter of Pichia pastoris. J Biosci Bioeng 92:585–589
Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B (1993) Specific binding sites in the alcR and alcA promoters of the ethanol regulon for the CREA repressor mediating carbon cataboiite repression in Aspergillus nidulans. Mol Microbiol 7:847–857
Leão-Helder AN, Krikken AM, Lunenborg MGJ, Kiel JA, Veenhuis M, van der Klei IJ (2004) Hansenula polymorpha Tup1p is important for peroxisome degradation. FEMS Yeast Res 4:789–794
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595
Li P, Anumanthan A, Gao XG, Ilangovan K, Suzara VV, Düzgüneş N, Renugopalakrishnan V (2007) Expression of recombinant proteins in Pichia pastoris. Appl Biochem Biotechnol 142:105–124
Lin-Cereghino GP, Godfrey L, de la Cruz BJ, Johnson S, Khuongsathiene S, Tolstorukov I, Yan M, Lin-Cereghino J, Veenhuis M, Subramani S, Cregg JM (2006) Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol Cell Biol 26:883–897
Marguerat S, Bähler J (2010) RNA-seq: from technology to biology. Cell Molr Life Sci 67:569–579
Mathieu M, Felenbok B (1994) The Aspergillus nidulans CREA protein mediates glucose repression of the ethanol regulon at various levels through competition with the ALCR-specific transactivator. EMBO J 13:4022–4027
Monastryska I, Kiel JAKW., Krikken AM, Komduur JA, Veenhuis M, van der Klei IJ (2005) The Hansenula polymorpha atg25 gene encodes a novel coiled-coil protein that is required for macropexophagy. Autophagy 1:92–100
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-SEq. Nat Methods 5:621–628
Nehlin JO, Ronne H (1990) Yeast MIG1 repressor is related to the mammalian early growth response and Wilms’ tumour finger proteins. EMBO J 9:2891–2898
Nehlin JO, Carlberg M, Ronne H (1991) Control of yeast GAL genes by MIG1 repressor: a transcriptional cascade in the glucose response. EMBO J 10:3373–3377
Ostling J, Carlberg M, Ronne H (1996) Functional domains in the Mig1 repressor. Mol Cell Biol 16:753–761
Ozimek P, Veenhuis M, van der Klei IJ (2005) Alcohol oxidase: a complex peroxisomal, oligomeric flavoprotein. FEMS yeast Res 5:975–983
Ozsolak F, Milos PM (2010) RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12:87–98
Sahu U, Krishna Rao K, Rangarajan PN (2014) Trm1p, a Zn(II)2Cys6-type transcription factor, is essential for the transcriptional activation of genes of methanol utilization pathway, in Pichia pastoris. Biochem Biophys Res Commun 451:158–164
Sasano Y, Yurimoto H, Kuriyama M, Sakai Y (2010) Trm2p-dependent derepression is essential for methanol-specific gene activation in the methylotrophic yeast Candida boidinii. FEMS Yeast Res 10:535–544
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108
Schüller HJ (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr genet 43:139–160
Schultz J, Carlson M (1987) Molecular analysis of SSN6, a gene functionally related to the SNF1 protein kinase of Saccharomyces cerevisiae. Mol Cell Biol 7:3637–3645
Sibirny AA (2016) Yeast peroxisomes: structure, functions and biotechnological opportunities. FEMS Yeast Res 16:fow038
Stasyk OG, Van Zutphen T, Ah Kang H, Stasyk OV, Veenhuis M, Sibirny AA (2007) The role of Hansenula polymorpha MIG1 homologues in catabolite repression and pexophagy. FEMS Yeast Res 7:1103–1113
Stasyk OG, Maidan MM, Stasyk V, Van Dijck P, Thevelein JM, Sibirny AA (2008) Identification of hexose transporter-like sensor HXS1 and functional hexose transporter HXT1 in the methylotrophic yeast Hansenula polymorpha. Eukaryot cell 7:735–746
Van der Klei IJ, Yurimoto H, Sakai Y, Veenhuis M (2006) The significance of peroxisomes in methanol metabolism in methylotrophic yeast. BBA-Mol Cell Res 1763:1453–1462
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034
Vogl T, Glieder A (2013) Regulation of Pichia pastoris promoters and its consequences for protein production. New Biotechnol 30:385–404
Walther K, Schüller HJ (2001) Adr1 and Cat8 synergistically activate the glucose-regulated alcohol dehydrogenase gene ADH2 of the yeast. Microbiology 147:2037–2044
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang X, Wang Q, Wang J, Bai P, Shi L, Shen W, Zhou M, Zhou X, Zhang Y, Cai M (2016) Mit1 transcription factor mediates methanol signaling and regulates alcohol oxidase 1 promoter in Pichia pastoris. J Biol Chem 291:6245–6261
Wang J, Wang X, Shi L, Qi F, Zhang P, Zhang Y, Zhou X, Song Z, Cai M (2017) Methanol-independent protein expression by AOX1 promoter with trans-acting elements engineering and glucose-glycerol-shift induction in Pichia pastoris. Sci Rep 7:41850
Wilhelm BT, Landry JR (2009) RNA-seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48:249–257
Yurimoto H, Oku M, Sakai Y (2011) Yeast methylotrophy: metabolism, gene regulation and peroxisome homeostasis. Int J Microbiol 2011:101298
Zhang P, Zhang W, Zhou X, Bai P, Cregg JM, Zhang Y (2010) Catabolite repression of AOX in Pichia pastoris is dependent on hexose transporter PpHxt1 and pexophagy. Appl Environ Microbiol 76:6108–6118
Acknowledgements
This work was supported by Shanghai Science and Technology Innovation Plan (17JC1402400); Fundamental Research Funds for the Central Universities (22A201514040) and Grants of Young and Middle-aged Leading Science and Technology Innovation Talents from Ministry of Science and Technology of China. The authors are grateful to Prof. James M. Cregg, BioGrammatics, Inc. and Keck Graduate Institute of Applied Life Science, for helpful suggestions. The authors also thank Shanghai Huaguan Biochip Co. Ltd. for the help of RNA-seq data analysis.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Lei Shi declares that she has no conflict of interest. Xiaolong Wang declares that he has no conflict of interest. Jinjia Wang declares that she has no conflict of interest. Ping Zhang declares that she has no conflict of interest. Fei Qi declares that he has no conflict of interest. Menghao Cai declares that he has no conflict of interest. Yuanxing Zhang declares that he has no conflict of interest. Xiangshan Zhou declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shi, L., Wang, X., Wang, J. et al. Transcriptome analysis of Δmig1Δmig2 mutant reveals their roles in methanol catabolism, peroxisome biogenesis and autophagy in methylotrophic yeast Pichia pastoris . Genes Genom 40, 399–412 (2018). https://doi.org/10.1007/s13258-017-0641-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-017-0641-5