
Abstract
The alternative splicing pattern of transcription units can be influenced by the genotype of a neighboring locus, which is termed splicing quantitative trait locus (sQTL). Here we report a comprehensive catalog of sQTLs discovered from the public RNA-seq and matched genotype datasets of three European ancestries. Each pair of RNA-seq and genotype dataset was analyzed with IVAS, a locally developed R/Bioconductor package for sQTL discovery. A meta-analysis was applied to the three result sets to reach a consensus of 2525 sQTLs (FDR < 0.05). Among them, nine independent sQTLs overlapped the known signals in the catalog of genome-wide association studies. Interestingly, six of these sQTLs are associated with the alternative exons, whose absence would hamper the protein function by omitting a critical/conserved domain. Altogether, we report the list of candidate sQTLs, and it might be useful for the explanations of trait-associated polymorphisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genome-wide association (GWA) studies have been a powerful method for identifying genetic components associated with traits or diseases (Manolio 2009). Until now, about 15,000 published trait-associated SNPs have been compiled in the catalog of published GWA studies (http://www.genome.gov/gwastudies). In spite of a great success of the studies, interpreting these genetic loci remains challenging, as bulk of them do not change amino acid sequence of a protein (Ioannidis et al. 2009). Recently, there have been several approaches that attempt to explain the genetic mechanism of these GWA signals. For example, there are some web services that accept a list of GWA signals and look up the overlapping epigenetic loci, the regulatory roles of which have been compiled by the epigenomic studies such as the ENCODE project. Another example is based on the observations that GWA signals may affect disease susceptibility by altering regulations of gene expression (ENCODE Project Consortium 2004; ENCODE Project Consortium 2012). Expression quantitative trait loci (eQTLs) influence gene expression levels, while splicing quantitative trait loci (sQTLs) alter alternative splicing (AS) patterns. Previous studies have accumulated a number of eQTLs, which have been helpful to explain GWAS signals (Boyle et al. 2012; Gong et al. 2016; Nicolae et al. 2010; Schadt et al. 2008). On the other hand, only a few studies have reported a genome-wide survey of sQTLs. Since changes in AS have been well known for causing various diseases and influence the susceptibility to complex diseases, sQTLs altering the AS pattern may affect the balance of expression of transcript isoforms and consequently influence on traits or diseases (Wang and Cooper 2007; E et al. 2013). As a bulk of GWAS signals is located in noncoding region such as intron, UTR, and promoter (Zhang and Lupski 2015), it is tempting to investigate whether some GWA signals can be interpreted as sQTLs. Recent development of high-throughput RNA sequencing (RNA-seq) using NGS has enabled more precise annotation and quantification of exons than the conventional exon array method. Consequently, RNA-seq has been become a powerful alternative method for the survey of genome-wide sQTLs (Katz et al. 2010). Here, we report candidate genome-wide sQTLs in three European populations (CEU, GBR, and FIN). First, in order to facilitate the identification of candidate sQTLs, we developed a user-friendly R/Bioconductor package, IVAS, which identify genetic variants affecting a specific exon based on a simple transcript expression data set in fragment per kilobases for million reads (FPKM) from RNA-seq and a genotype data set. Using the package, we identified sQTLs with each of the published RNA-seq datasets and genotype datasets generated from the lymphoblastoid cell line in the populations of the 1000 Genomes Project (Consortium et al. 2010). The results of the individual studies were then combined by a meta-analysis in order to reduce the heterogeneity among the studies and to improve the statistical power (Begum et al. 2012; Teslovich et al. 2010). The meta-analyzed sQTLs comprise a comprehensive collection of genome-wide sQTLs, which may be a useful resource in understanding the genetic mechanisms of GWA signals.
Materials and methods
Study samples and data preprocessing
The matched RNA-seq and genotype data used in this study have been downloaded from the Geuvadis RNA Sequencing Project (http://www.geuvadis.org/web/geuvadis/RNA-seqproject) and the 1000 Genomes Project (Consortium et al. 2010), respectively. The RNA-seq project measured RNA expressions from the lymphoblastoid cell lines of the three European populations (78 CEU, 89 FIN, and 85 GBR individuals) included in the 1000 Genomes Project.
The downloaded RNA-seq quantification data include 183,086 transcript FPKM values. Among them, we considered only genes with at least two isoforms. Furthermore, we selected 4712,172 SNPs out of 38,187,571 markers in the genotype data by minor allele frequency (MAF) > 0.1. Among them, we studied 196,853, 194,237, and 198,950 SNPs located in the alternative exons and their flanking introns in CEU, FIN and GBR populations, respectively.
Meta-analysis of three European populations
In order to improve statistical power and decrease heterogeneous associations among the three populations, the individual IVAS results were combined using METAL, a meta-analysis software (Willer et al. 2010). We selected SNPs with heterogeneity P-value > 0.85, FDR P-value based on the meta-analysis <0.05, and Z-score > 2.75.
Estimation of expression ratio for alternative spliced exons
The relative expression ratio between included and skipped exons is used for evaluating the exon inclusion. For a specific exon j, the relative expression ratio, Ψj, is as follows:
The Xij and Xsj are transcript expressions with and without exon j, respectively (Supplementary Fig. 2). We implemented this method in a user-friendly and convenient R/Bioconductor package, IVAS.
General process of IVAS
The package is written in the R programming language and has dependency on other packages of R/Bioconductor. To run this package, two experimental datasets (an expression and genotype data frame) are required. In addition, it also needs a data frame of marker positions and a general transfer format (GTF) file of reference transcript models. Based on the reference transcript models of a given gene, IVAS identifies exons that are alternatively included among the transcript isoforms. For each exon of interest, IVAS divides the isoforms into two groups: transcripts with and without it. The group totals of the transcript expression levels are calculated, and their ratio is taken as an indicator of alternative exon skipping (Supplementary Fig. 2). Finally, IVAS looks for statistically significant correlations between genotypes and the ratio values with previously used statistical models for sQTLs. The user manual including installation, implementation, and tutorials is provided as a vignette in the package. This package is available at http://bioconductor.org/packages/release/bioc/html/IVAS.html.
Results
Identification of sQTLs in the CEU population using IVAS
We used the locally developed IVAS package (see “Materials and methods”) section for the identification of sQTLs. Initially to validate the IVAS performance, we analyzed the lymphoblastoid cell line samples in CEU population of the 1000 Genomes Project (Consortium et al. 2010), and compared the results with those by other tools with the same dataset. The CEU genotype and the RNA-seq dataset were downloaded from the Geuvadis RNA sequencing project website (http://www.geuvadis.org/web/geuvadis/RNA-seq project). The RNA-seq dataset contains FPKM values of 183,086 unique transcripts, and the genotype dataset contains 38,187,571 SNP markers. The genotype data were filtered by MAF > 0.1, resulting in 4712,172 SNP markers. Furthermore, we considered only genes with at least two AS isoforms, as well as only SNPs located within the alternative exons and their flanking introns. The filtered genotype and expression data sets comprising 196,853 SNPs and 6279 genes were analyzed with IVAS. As a result, we discovered 562 sQTLs affecting 74 alternative exons at a FDR of 1 %. In order to show that our result is reliable, we compared our result with other tools. There are two previously published tools, GLiMMPS (Zhao et al. 2013) and sQTLseekeR (Monlong et al. 2014), for identifying sQTLs. We compared the result with GLiMMPS, a tool that estimates the expression level based on junction read counts. GLiMMPS could not test 23 out of 74 sQTL target exons that had been identified by IVAS in the CEU population. Those 23 target exons lacked sufficient junction reads to be analyzed by GLiMMPS, while they had enough exon reads to be identified by IVAS (Supplementary Fig. 1). Of the remaining 51 exons (associated with 478 sQTLs), GLiMMPS identified 36 (associated with 445 sQTLs) as being significant (P < 0.05). The second tool we compared against was sQTLseekeR, which investigates multivariate comparison of all isoforms expression values individually in the transcript level. Unlike GLiMMPS, only one SNP from its result overlapped with the significant SNPs from our result. The detailed comparisons are given in Supplementary Figs. 3 and 4.
Meta-analysis of three European populations
Upon a successful validation of our IVAS tool with the CEU dataset, we analyzed two additional European datasets, namely GBR and FIN populations, also available from the Geuvadis project (http://www.geuvadis.org/web/geuvadis/RNA-seq project). The IVAS runs, comprising an average of 196,853 SNPs and 6279 genes in the studied populations, identified 562, 492, and 485 candidate sQTLs and 74, 60, and 71 alternatively spliced target exons at a FDR of 1 % in the CEU, GBR and FIN populations, respectively (Supplementary Table 1). The high portion of candidate sQTLs was shared across three test populations (Fig. 1). If we focus on CEU and GBR, which are known to be closer to each other than FIN (Olalde et al. 2014), the sharing rate is about 74 %. All those three results were combined by a meta-analysis approach, in order to improve statistical power in discovering true sQTLs and to reduce heterogeneous calls among the three populations. The meta-analysis was performed using METAL (Willer et al. 2010), resulting in a comprehensive list of 2525 statistically significant loci (P (heterogeneity) > 0.85 (meaning highly homogenous calls), FDR (after the meta-analysis) < 0.05, and Z-score > 2.75). The result is found in Supplementary Table 2.

The sharing of sQTLs between populations. The sharing rate, ij at the column i and row j, is defined as follows: Cij = |Xi ∩ Xj|/|Xi| (i ∈ {CEU, FIN, GBR}) where X i is the set of sQTLs identified in the population i
Interpretation of trait-associated SNPs with sQTLs
Here we demonstrate the utility of sQTLs in interpreting known GWAS signals. The published trait-associated SNPs were downloaded from the catalog of published GWA Studies (http://www.genome.gov/gwastudies). For each of the sQTLs that have been compiled through the meta-analysis, the neighboring SNPs with high linkage disequilibrium (r2 > 0.8) were compiled from dbSNP, expanding the genomic loci. We then looked for those candidate loci that overlapped with the list of the GWAS catalog SNPs. As a result, nine independent sQTLs overlapped the trait-associated SNPs in the GWAS catalog, and their alternatively spliced target exons were annotated using the ensembl genome browser (Table 1). Interestingly, in six cases, the alternative exon skipping cause a loss of either a key functional domain or a coding feature conserved in vertebrates. The remaining three cases involve skipping exons in 5′-UTR regions that are conserved in mammals.
Discussion
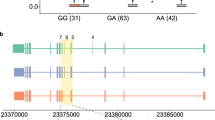
We used a locally developed R/Bioconductor package, IVAS, a convenient and user-friendly tool for the discovery of sQTL, genetic variants affecting alternative splicing. To our knowledge, there have been two published software tools, GLiMMPS and sQTLseekeR, for sQTL discovery. GLiMMPS counts the reads spanning splice junctions to estimate the expression level of the sQTL target exon. Unless enough junction reads are available, this method may miss the sQTL target exons. On the other hand, sQTLseekeR overcomes such an issue by comparing the expression levels of the whole transcripts given in terms of so-called FPKM values. However, this tool does not pinpoint the alternative exons that are differentially expressed. Our tool, IVAS, decouples the transcript FPKM values into the expression level of a specific target exon. Using IVAS, we report candidate sQTLs in three European populations. The availability of three closely related population datasets allowed us to infer study variations. The population structure analysis based on genotype differences has reported that while these populations are part of the European cluster that are distinct from either African or Asian populations, CEU and GBR are closer to each other than to FIN (Olalde et al. 2014). The three separate lists of candidate sQTLs from our IVAS runs also reflected this population structure: the generally high sharing among all three populations and even higher overlap between CEU and GBR, a closer pair, than either with FIN, a remoter neighbor (Fig. 1). This indicates that our sQTL analysis captures some genetic components in transcription regulation rather than manifests arbitrary spurious statistical associations. The fact that the three results are comparable to one another prompted us to do an integrative approach to compile a unified list of candidate sQTLs. The Fig. 2b shows exemplary boxplots of a sQTL, rs17418283, in three datasets that show the correlation between its genotype and an expression level of an MCTP1 exon. When the sharing of sQTLs is high as in this example, the meta-analysis can improve the sensitivity of detecting sQTLs. The inconsistency among the populations can be inferred by the heterogeneity calculation as a part of the meta-analysis, and the P (heterogeneity) cutoff can be used to remove inconsistent results. We used a filter of P (heterogeneity) > 0.85 in order to minimize the inconsistent associations.

The effect of 18th-exon skipping in MCTP1. a The SNP rs17418283, an intronic variant of MCTP1, is associated with the 18th-exon skipping in MCTP1. b The distribution of expression ratio of the exon in each genotype of rs17418232. The higher y-value corresponds to more exon inclusion. c The protein structure of MCTP1 was predicted by RaptorX structure prediction tool, a Web based prediction tool for protein structure and function (Kallberg et al. 2014). The 18th-exon skipping leads to missing one of the transmembrane helices (the blue arrow)
As the sQTLs affect the balance of expression of transcripts by changing AS patterns, their functional consequences can contribute to the phenotypic variation or disease susceptibility. In that regard, a comprehensive list of sQTLs can be a useful resource for the underpinning of genetic associations of various traits or diseases. For example, MCTP1 encodes a protein having two C-terminal transmembrane domains. An intronic variant in MCTP1, rs17418283, was reported as having an association with bipolar disease (Scott et al. 2009). However, its molecular underpinning has been elusive. Our study identified the rs17418283 was associated with the alternative skipping event of the 18th-exon, resulting in the loss of one of the transmembrane helices (Fig. 2). It turned out that the allelic directions of the risk allele of our result (enhancing the exon skipping) and the GWA signal (promoting bipolar disease) were opposite. High intracellular accumulation of calcium ion was reported in bipolar disease. Since MCTP1 protein can bind calcium ion and plays a role in calcium-mediated signaling, it is tempting to speculate that the canonical form of MCTP1 may be one of the risk factors of bipolar disease. Further biochemical and functional studies of the alternative MCTP1 protein isoforms may help elucidate its role in bipolar disease pathogenicity. Currently only a small portion of the GWAS Catalog can be explained in terms of sQTLs. As more GWAS signals are cataloged in the coming years, our current list of candidate sQTLs may find more utility. Moreover, more candidate sQTLs can be discovered by studying a variety of tissues other than lymphoblastoid cell lines in population level. Such a comprehensive list of sQTLs would be a useful resource in functional genomics.
References
Allen HL, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S et al (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467:832–838
Begum F, Ghosh D, Tseng GC, Feingold E (2012) Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res 40:3777–3784
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S et al (2012) Annotation of functional variation in personal genomes using Regulome DB. Genome Res 22:1790–1797
Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, Butte NF (2012) Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One 7:e51954
Consortium TGP, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population scale sequencing. Nature 467:1061–1073. doi:10.1038/nature09534
Deng X, Sabino EC, Cunha-Neto E, Ribeiro AL, Ianni B, Mady C, Busch MP, Seielstad M (2013) Genome wide association study (GWAS) of Chagas cardiomyopathy in Trypanosoma cruzi seropositive subjects. PLoS One 8:e79629
E Z, Wang L, Zhou J (2013) Splicing and alternative splicing in rice and humans. BMB Rep 46:439–447
ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) project. Science 306:636–640
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Goes FS, Hamshere ML, Seifuddin F, Pirooznia M, Belmonte-Mahon P, Breuer R, Schulze T, Nothen M, Cichon S, Rietschel M et al (2012) Genome-wide association of mood-incongruent psychotic bipolar disorder. Transl Psychiatry 2:e180
Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P et al (2008) Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA 105:4340–4345
Gong C, Xu Y, Fan Y, Liu X, Xiong C, He L, Liu C, Rao S, Xia W, Ding L et al (2016) Functional mechanisms for diabetic nephropathy-associated genetic variants. Genes Genom. doi:10.1007/s13258-016-0415-5
Ioannidis JP, Thomas G, Daly MJ (2009) Validating, augmenting and refining genome-wide association signals. Nat Rev Genet 10:318–329
Kallberg M, Margaryan G, Wang S, Ma J, Xu J (2014) RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol 1137:17–27
Katz Y, Wang ET, Airoldi EM, Burge CB (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7:1009–1015
Manolio TA (2009) Cohort studies and the genetics of complex disease. Nat Genet 41:5–6
McDonough CW, Palmer ND, Hicks PJ, Roh BH, An SS, Cooke JN, Hester JM, Wing MR, Bostrom MA, Rudock ME et al (2011) A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int 79:563–572
Monlong J, Calvo M, Ferreira PG, Guigo R (2014) Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat Commun 5:4698
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6:e1000888
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S et al (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376–381
Olalde I, Sanchez-Quinto F, Datta D, Marigorta UM, Chiang CW, Rodriguez JA, Fernandez-Callejo M, Gonzalez I, Montfort M, Matas-Lalueza L et al (2014) Genomic analysis of the blood attributed to Louis XVI (1754–1793), king of France. Sci Rep 4:4666. doi:10.1038/srep04666
Parsa A, Chang YP, Kelly RJ, Corretti MC, Ryan KA, Robinson SW, Gottlieb SS, Kardia SL, Shuldiner AR, Liggett SB (2011) Hypertrophy-associated polymorphisms ascertained in a founder cohort applied to heart failure risk and mortality. Clin Transl Sci 4:17–23
Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C et al (2008) Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6:e107
Scott LJ, Muglia P, Kong XQ, Guan W, Flickinger M, Upmanyu R, Tozzi F, Li JZ, Burmeister M, Absher D et al (2009) Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc Natl Acad Sci USA 106:7501–7506
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Wang GS, Cooper TA (2007) Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8:749–761
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191
Zhang F, Lupski JR (2015) Non-coding genetic variants in human disease. Hum Mol Genet 24:R102–R110
Zhao K, Lu ZX, Park JW, Zhou Q, Xing Y (2013) GLiMMPS: robust statistical model for regulatory variation of alternative splicing using RNA-seq data. Genome Biol 14:R74
Acknowledgments
This work was supported in part by the Industrial Strategic Technology Development Program, 10,040,231, “Bioinformatics platform development for next-generation bioinformation analysis,” funded by the Ministry of trade, industry & energy (MOTIE, Korea). This work was also supported by the National Research Foundation, funded by the Ministry of Science, ICT & Future Planning, Korea (NRF-2012M3A9D1054705).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Seonggyun Han, Hyeim Jung, Kichan Lee, Hyunho Kim and Sangsoo Kim declare no conflict of interest.
Human and animal rights
This article does not contain any studies with human subjects or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Han, S., Jung, H., Lee, K. et al. Genome wide discovery of genetic variants affecting alternative splicing patterns in human using bioinformatics method. Genes Genom 39, 453–459 (2017). https://doi.org/10.1007/s13258-016-0466-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-016-0466-7