
Abstract
Variation is the primary source for plant and animal breeding and evolution, and hence, the detection of variation is an integral part of genetics, breeding, and ecology. Various molecular marker systems have been developed to detect genetic variation. They detect sequence variations (e.g., restriction fragment length polymorphism, randomly amplified polymorphic DNA, amplified fragment length polymorphism) or simple sequence motif variations. However, a large portion of genomic variation is derived from the transposition of transposable elements (TEs), which are major denizens of most eukaryote genomes. Therefore, molecular markers derived from TEs are valuable resources for dissecting genomes in plants and animals. Because class I retrotransposons transpose by a “copy-and-paste” semi-conservative manner, retrotransposon-based markers (e.g., Inter-retrotransposon amplified polymorphism, retrotransposon-microsatellite amplified polymorphism, sequence-specific amplified polymorphism) can reveal highly accurate phylogenetic relationships among related taxa as well as among accessions within a species. Transposon display based on class II DNA transposons has also been used in various genetics fields. A large amount of fairly accurate genome sequences are now being generated, and computational biology allows us to mine the TEs on a genome-wide scale. Thus, TE-based molecular markers are adding another venue to the other marker systems used for the molecular dissection of genomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The analysis of genetic variation has been an integral part of plant genetics, breeding, and ecology. Detecting genetic variation is a critical step in the analysis of polymorphisms, which were described as early as the Biblical record of Jacob’s lamb polymorphisms. Gregor Mendel drew inferences of genetic inheritance by analyzing seven polymorphic morphological traits in pea breeding experiments. However, the number of discernable morphological traits is not extensive, and environmental effects on morphological variation can restrict the utilization of morphological traits in modern genetic analyses, which require a large number of unbiased genetic markers for various purposes in crop and animal breeding, germplasm management, conservation ecology, and population genetics. Genetic markers are associated non-randomly with variation in natural or segregating populations. With the advancement of molecular genetic methodologies, various protein and DNA markers associated with genetic variation have been developed. Mutations are the primary sources of diversity and, thus, evolution. Transposable elements (TEs), common denizens of eukaryotic genomes, account for a large portion of the genetic variation in plants and animals. This review summarizes the molecular markers, TEs, and TE-derived molecular markers used in genetic analyses.
The development of molecular markers
The detection of variation by markers based on protein polymorphisms was adopted in the 1960s (Hubby and Lewontin 1966). The easy scoring of allozyme variation by electrophoresis supplemented the use of morphological markers during the 1970s and 1980s (Ganapathy and Scandalios 1973; Tanksley 1983). However, the limited number of detectable isozymes, tissue specificity, and environmental effects on isozyme expression were drawbacks to the use of isozymes for large-scale genetic analyses (Tanksley 1983). A method of detecting DNA polymorphism in lieu of using isozyme variation was developed in 1980 (Botstein et al. 1980). Restriction fragment length polymorphism (RFLP) assays can detect restriction-site variation in genome sequences by hybridizing molecular probes in a Southern blot analysis (Botstein et al. 1980). Because molecular probes can be developed from different sources of cDNAs and genomic clones, RFLP promised to detect a large amount of molecular variation (Botstein et al. 1980; Tanksley et al. 1989; Prince and Tanksley 1992). RFLP markers were popular during the 1980s and 1990s for genetic mapping, tagging agronomic traits, and demographic analyses of plant populations (reviews available in Tanksley et al. 1989; Bachmann 1994, references therein). Regardless of the advantages, the technical difficulty and requirement for large amounts of relatively pure genomic DNAs limited the routine utilization of RFLPs in practical applications and fieldwork.
The advent of polymerase chain reaction (PCR) made diverse, PCR-based marker systems possible. The first PCR-based marker system was randomly amplified polymorphic DNAs (RAPDs) (Willams et al. 1990). The RAPD technique is very simple, employing a short arbitrary primer (usually a decamer) in a PCR; but it has the major drawback of low reproducibility due to low annealing temperatures in the PCRs. In the mid-1990s, amplified fragment length polymorphism (AFLP) was developed to detect restriction-site variation by PCR (Vos et al. 1995). It employs restriction digestions using two different restriction enzymes and the ligation of adaptors to the restriction sites for PCR amplification, which allows for complex mixtures of amplified fragments. AFLP combines the reproducibility of RFLP with the simple PCR of RAPD. AFLP can detect many restriction-site variations with simple PCR reactions. Both RAPD and AFLP are dominant markers and do not require sequence information for designing the primers. The simple sequence repeat polymorphism (SSRP) (or microsatellite polymorphism) system was developed in the mid-1990s. It employs flanking primers to amplify simple sequence repeats (SSRs), which are highly abundant in eukaryotic genomes. It is the preferred molecular marker system in plant genetics and ecology research due to the high abundance, high reproducibility, and high allelic variation of the SSRPs as well as the simplicity of the technique (Ellergren 2004, references therein). However, SSRPs require sequence information to design the flanking primers that amplify the SSR motifs (Tauts and Renz 1984). Thus, although the SSRP system has many merits over other marker systems, its applicability to minor or neglected crops or plants is limited due to the paucity of DNA sequence information (Park et al. 2009).
Advances in genome sequencing techniques have enabled many innovative techniques for genetic marker development. The large amount of accurate sequences has allowed the identification of single nucleotide polymorphisms (SNPs) in many plant species (Ganal et al. 2009; Varshney et al. 2009). Array technologies have allowed the massive, genome-wide detection of SNPs and the analysis of genetic diversity in plants (Borevitz et al. 2003; Singer et al. 2006; Kumar et al. 2007). Diversity array technique (DArT) was a similar technique using arrays, but offered a low-cost high throughput analysis without sequence information in genome-wide genotyping (Jaccoud et al. 2001). Because plant genomes carry abundant repetitive sequences, and they carry scattered regions that were duplicated in ancient tetraploids, the SNP mining of genome sequences in plants is challenging (Mammadov et al. 2012). Transcriptome sequencing can identify SNPs in an inexpensive way with a low frequency of spurious false-positive or false-negative SNPs (Chagné et al. 2008). Sequencing the gene-enriched library derived from methylation-sensitive restriction fragments also minimizes repetitive DNA sequencing to improve SNP identification (Gore et al. 2009).
Transposable elements
TEs are mobile genetic elements that are found with high copy numbers in almost all eukaryotes (Wicker et al. 2007; Schulman and Wicker 2013). The transposition of TEs can generate genome plasticity by inducing various chromosomal mutations and allelic diversity (Fedoroff 2013; Oliver and Greene 2009; Oliver et al. 2013). Based on the transposition mechanisms, TEs are conventionally classified into two classes: class I TEs and class II TEs (Finnegan 1989). Class I TEs are retrotransposons that move in a semi-conservative “copy-and-paste” manner via RNA intermediates. Class II TEs are DNA transposons that transpose via DNA intermediates.
The original copy of a class I retrotransposon is transcribed into mRNA that is then reverse transcribed by the retrotransposon-encoded reverse transcriptase. The resulting DNA copy is then inserted into a new location while the original copy is retained at the original position, which results in genome expansion (Fedoroff and Bennetzen 2013; Lee and Kim, 2014). The class I retrotransposons are subdivided into two subfamilies: long terminal repeat (LTR) retrotransposons and non-LTR retrotransposons. Upon integration, the LTR retrotransposons often produce target site duplications (TSDs) of 4–6 bp. The LTR retrotransposons are more prevalent in plant genomes and carry two open reading frames (ORFs), GAG and POL (Voytas and Boeke 2002; Levin 2002). GAG encodes a protein for replication, and POL encodes a multi-protein comprising protease (PR), integrase (INT), reverse transcriptase (RT), and RNase H (RH). Depending on the order of the genes encoded, the LTR retrotransposons are further classified into Ty1-copia and Ty3-gypsy retrotransposons (Fig. 1). The gene order of Ty1-copia retrotransposons is PR-INT-RT-RH, whereas that of Ty3-gypsy retrotransposons is PR-RT-INT-RH. Although several types of non-LTR retrotransposons have been reported, long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) are the typical non-LTR retrotransposons. LINE-1, a typical LINE in mammals, carries two ORFs; a short ORF encodes a nucleic acid binding protein, and a long ORF encodes a protein with endonuclease and reverse transcriptase activities (Moran and Gilbert 2002). LINEs are usually 4–6 kb in size and mostly found in mammals, except for Del-2. Del-2 is highly abundant in the monocotyledonous Lilium species (Leeton and Smyth 1993). SINEs are small in size (80–150 bp) and highly abundant in animal genomes. The Alu element, the best-known SINE, is present in up to 500,000 copies in the human genome (Rowold and Herrara 2000). The SINEs are elusive in their origin but are presumed to be derived from various polymerase III transcripts, including 7SL or any of several tRNA genes (Sakamoto and Okada 1985).

Structure of the Ty1-copia and Ty3-gypsy retrotransposons. Both LTR retrotransposons have long terminal repeats at both ends. The gene order at the Pol region is different between the Ty1-copia and Ty3-gypsy elements. PBS primer binding site, PPT poly purine track, RH RNase H, INT integrase, RT reverse transcriptase
Class II TEs are DNA transposons that transpose by single-stranded or double-stranded DNA intermediates (Craig et al. 2002). The class II TEs are subdivided into subclass I, the terminal inverted repeat (TIR) transposons, and subclass II, the non-TIR transposons (Wicker et al. 2007; Schulman and Wicker 2013). TIR transposons transpose by a conservative “cut-and-paste” mechanism. The TIRs form a fold-back structure, which is excised by a transposase encoded by the transposon itself. The excised double-stranded DNA reinserts elsewhere in the genome. Based on the TIR sequences and the TSD sizes, the TIR transposons are further classified into nine superfamilies. Six superfamilies (Tc1-Mariner, hAT, Mutator, P, PIF-Harbinger, and CACTA) are found in plants. The TIR transposons increase their copy number by transposition from replicated chromatids to unreplicated chromosomes during DNA replication. Alternatively, gap repair of the donor site after excision can also increase the copy number of TIR transposons. However, the TIR transposons do not attain copy numbers as high as those of the LTR-retrotransposons. Miniature inverted-repeat transposable elements (MITEs) are TIR transposons (Bureau and Wessler 1992). MITEs are small in size and do not carry an ORF. Also, they are present in very high copy numbers in plant genomes. A careful comparison between the MITEs and the other TIR transposons revealed that Stowaway MITEs are derived from the Tc1/mariner superfamily, and Tourist MITEs are derived from the PIF/Harbinger superfamily (Feschotte et al. 2002a, b). The Helitron superfamily of subclass-2 non-TIR DNA transposons is present in plants (Wicker et al. 2007). Unlike the double-stranded cleavage used for integration by the subclass-1 DNA transposons, the subclass-2 non-TIR DNA transposons undergo transposition without double-stranded breakage. The Helitron elements appear to replicate via a rolling-circle mechanism involving the displacement of only one DNA strand during transposition, and they do not produce TSDs (Kapitonov and Jurka 2001). Helitrons are present in high copy numbers and capture pseudogene fragments in maize, which contributes to the high genomic diversity of maize (Du et al. 2009).
Molecular markers derived from transposable elements
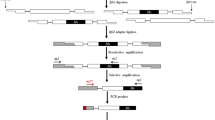
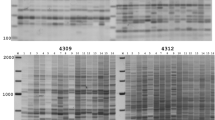
TEs have been utilized as genetic markers because of their genome-wide distributions (Kalendar et al. 2011; Poczai et al. 2013; Bonchev and Parisod 2013). TE integration into functional genes often results in null alleles, and the excision of TEs from such sites could restore allelic function. Variable phenotypes derived from TE mobilization have provided efficient genetic and molecular tools for gene discovery and isolation through forward and reverse genetic strategies (Bensen et al. 1995; Das and Martienssen 1995; Kumar and Hirochika 2001). The ubiquity, high copy numbers, and genome-wide distributions of TEs have rendered genome-wide genetic markers out of both the class I and the class II TEs (Feschotte et al. 2002a, b; Huang et al. 2008; Zhuang et al. 2014). Transposon-display (TD) is a modified AFLP technique used to detect TE insertion polymorphisms (Korswagen et al. 1996) (Fig. 2). While the AFLP protocol detects restriction site variation, TD detects the presence/absence of TEs, which have been utilized in various fields of genetics, plant breeding, and ecology (Le and Bureau 2004; Grzebelus 2006; Lockton et al. 2008; Kalendar et al. 2011). Comparisons between AFLP-derived and retrotransposon-derived markers showed that the retrotransposon-derived markers were more informative in many studies of genetic diversity and demography among plant species (Tam et al. 2005; Kalendar et al. 2011). In our unpublished data from 23 different ecotypes of Arabidopsis thaliana, the observed heterozygosity and genetic diversity measured by class II transposon CACTA markers were higher than those measured by AFLP (Fig. 3). Table 1 illustrate the comparison of molecular marker systems of isozyme, mutation based markers, marker system using array technique, and TE-based markers.

Transposon display (TD). Genomic DNA is restriction-digested with Mse I, which recognizes TTAA to make a TA overhang at the cutting site. An adaptor for the restriction site is then ligated. Pre-amplification is carried out with a primer (MseI + 0; no selective nucleotides) complementary to the adaptor and a primer (P1) complementary to the internal site of the transposon. Then, selective amplification is carried out with an MseI adaptor primer with selective nucleotides (MseI + 3N) and a primer (P2) complementary to an internal sequence that is complementary to the terminal inverted repeat and the P1 site. In the diagram, the filled arrowheads at each end are the terminal inverted repeats (TIRs). The logic of the sequence-specific amplified polymorphism (SSAP) is the same as that of the TD except that SSAP utilizes LTR retrotransposons instead of DNA transposons

Fingerprinting profiles of ten ecotypes of Arabidopsis thaliana by AFLP (a) and CACTA-TD (b). The profiles revealed that the transposition based variations are higher than restriction site variations
Class I retrotransposon-derived molecular markers
Class I retrotransposons are highly abundant and widely distributed in eukaryotes, particularly in plant genomes. The retrotransposons transpose to new sites after transcription so that the original copy remains in its site in the genome (Finnegan 1989). This unidirectional transposition of the class I retrotransposons can provide highly informative utilities in phylogenetic analyses (Vitte et al. 2004; Grzebelus 2006; Jing et al. 2010). LTR retrotransposons are more prevalent than non-LTR retrotransposons in plants. Their chromosomal locations are either clustered in pericentric or intercalary heterochromatic regions or dispersed throughout the genome, which makes them suitable for developing PCR-based markers. Primers are usually designed from the LTRs near the insertion sites. The LTR subdomain sequences are conserved within retrotransposon families but are different between families, so many insertion polymorphisms within a retrotransposon family can be retrieved by a single PCR amplification using a primer that is complementary to the LTR. The LTR retrotransposon-derived markers are all dominant markers, so they cannot distinguish between insertion homozygotes and insertion heterozygotes. Although numerous retrotransposon-based marker systems were contrived, only the four most frequently used systems are covered in this review (Fig. 4).

LTR retrotransposon-based marker systems. a IRAP (inter-retrotransposon amplified polymorphism). IRAP amplifies between LTR transposons using a set of primers that are complementary to the 3′-end of LTRs to be outbound. LTRs are either head-to-tail (top) or head-to-head (bottom). If adjacent LTR-retrotransposons are same elements, a single primer can amplify the spacers. If they are different elements, two different primers are needed to amply. b REMAP (retrotransposon-microsatellite amplified polymorphism). REMAP amplifies between LTR-retrotransposon and adjacent microsatellites. PCR carries out with a primer complementary to the 5′- or 3′-end of LTR and a primer with a simple sequence motif plus selective nucleotides [e.g., (CGG)5G, (ATT)5C]. For multicopy elements in complex genomes, selective nucleotides can be added to the LTR primer to reduce amplified fragments. c RBIP (retrotransposon based insertion polymorphism). Unlike other retrotransposon-based markers, RBIP detects a single locus of presence/absence of retrotransposon. It utilizes three primers for each locus. Two primers (P1 and P3) are flanking primers inward to the locus. Another primer (P2) is an out-bound primer from the transposon. If LTR-retrotransposon is present at the locus, the P1 + P2 primer sets can amplify the target site, but the P1 + P3 primer sets do not yield products because the distance between two primers is beyond the PCR amplification. If the locus is empty, P1 + P2 do not yield amplification, but the P1 + P3 do yield amplification
Inter-retrotransposon amplified polymorphism (IRAP)
The IRAP system amplifies the sequences between two adjacent LTR retrotransposons by utilizing primers that are complementary to the 3′ end of the LTR sequence (Kalendar et al. 1999). It can amplify the spacer sequences either between LTR retrotransposons of the same lineage by a single primer or between LTR retrotransposons of different lineages by a set of primers derived from each lineage of LTR sequences. Because LTRs are direct repeats, the primers facing outward from the 3′ end of the right LTR can also prime the 3′ end of left LTR, which amplifies the interior sequences of the LTR retrotransposon. The interior amplification can be avoided by designing the primers to overlap bases at the 3′ end that do not match the LTR-interior interior junction (Kalendar et al. 2011). The LTR sequences between adjacent retrotransposons are arranged (a) head-to-head, (b) tail-to-tail, or (c) head-to-tail (Poczai et al. 2013). If the arrangement between two identical tandem duplicate LTR retrotransposons is either head-to-head or tail-to-tail, a single primer can amplify the spacer. If the adjacent retrotransposons are from different lineages (which is usually the case), two different primers derived from each LTR sequence are needed to amplify the IRAP. Each IRAP reaction produces multiple amplicons ranging in size from 300 to 3,000 bp (Branco et al. 2007; Fan et al. 2014). A major advantage of the IRAP technique is its experimental simplicity, because all that is needed in IRAP is a simple PCR and subsequent electrophoresis. Vukich et al. (2009) analyzed the genetic variability among Helianthus species using IRAP and detected a species-specific insertion of a Copia-like element, Helicopia, and distinct fingerprints distinguishing the annual and perennial Helianthus species. More recently, Fan et al. (2014) utilized the IRAP technique to dissect the genetic diversity of the masson pine (Pinus massoniana), which revealed very high genetic diversity in that gymnosperm species. They detected the expression of reverse transposases from both the Copia-type and the Gypsy-type retrotransposons by exposing the plants to various hormones and environmental stresses, but no changes were detected in the IRAP fingerprinting among the masson pine specimens.
Retrotransposon-microsatellite amplified polymorphism (REMAP)
The REMAP protocol is similar to that of IRAP. REMAP utilizes microsatellites (or SSRs) in conjunction with LTR-specific primers in PCRs (Kalendar et al. 1999; Kalendar and Schulman 2006). Microsatellites are short, SSRs that are highly redundant and polymorphic in eukaryotic genomes (Temnykh et al. 2001; Park et al. 2009). The REMAP PCR uses primers for microsatellite loci containing the repeat motif plus an additional anchoring nucleotide at the 3′ end [e.g., (CA)nA, (GC)nC] to avoid slippage of the primer between the individual SSR motifs. REMAP detects multiple loci that are similar in size and number distribution to those detected by IRAP. Both IRAP and REMAP reveal very broad polymorphic profiles among different genotypes within species as well as between species within a genus. Kalendar et al. (1999) analyzed the genomic distributions of the BARE-1 retrotransposon in 15 barley varieties (Hordeum vulgare L.) using the IRAP and REMAP protocols, which allowed a clear distinction of the varieties. Moreover, the same primer combinations were easily transferable for the fingerprinting of related species within the genus. Similarly, Branco et al. (2007) demonstrated the amenability of both IRAP and REMAP to the assessment of genetic similarity among rice varieties, and they presented the results of the differentiation of the Brazilian and Japanese rice varieties.
Sequence-specific amplified polymorphism (SSAP)
SSAP is a modified AFLP protocol. While AFLP requires no a priori sequence information and employs two restriction enzymes and adaptor systems to amplify the restriction-digested fragments (Vos et al. 1995), SSAP requires a priori transposon sequence information (Syed and Flavell 2007). SSAP employs a single restriction digestion and the ligation of an adaptor to the restriction site. Then, a primer complementary to the 3′ end of the LTR and a primer complementary to the adaptor sequence are used for the SSAP PCR. Genomic DNA is digested with a restriction enzyme, and adaptors are ligated to the restricted end. Then, a primer complementary to the adaptor and a primer complementary to the 3′ end of the LTR sequence are employed in the PCR. If the elements are low in copy number, the first amplification may yield discernable bands in denaturing polyacrylamide gel electrophoresis. If the first amplification produces too many bands to read, the second amplification with selective nucleotides at the ends of both primers can reduce the number of bands (Poczai et al. 2013). The SSAP protocol is basically the same as the TD employed for the amplification of the class II DNA transposons (Fig. 2). In the case of the class II transposons, the TD protocols were named for the particular transposon class that was incorporated (e.g., MITE-TD, CACTA-TD). SSAP usually produces a large number of amplicons that represent the TE insertion sites, but mutations at the restriction sites may also yield polymorphisms in SSAP assays (Petit et al. 2010). The retrotransposon insertion sites identified by SSAP are highly reproducible and produce multiple fragments that cover the whole genome, which are suitable for examining the LTR-retrotransposon insertions at a specific level. Moisy et al. (2008) surveyed 10 Ty1-copia-like retrotransposon families in the Vitis genus and showed that most of the scorable bands were polymorphic and that only a few insertion sites were fixed in the accessions surveyed. Genomic shocks, including biological or abiological stresses, and polyploidization can induce the mobilization of both class I and class II TEs (McClintock 1984; reference). Woodrow et al. (2010) demonstrated that the retrotransposition of Ty1-copia-like elements plays important roles in defense responses to environmental stresses in tetraploid wheat. Allopolyploidy is a major driving force in plant evolution, inducing rapid structural changes in hybrid genomes; TEs are major components of those changes (Liu and Wendel 2000; Kashkush et al. 2002; Josefesson et al. 2006; Parisod and Senerchia 2012). Petit et al. (2010) analyzed retrotransposon mobilization in synthetic allotetraploid tobacco by SSAP analysis of a Copia-type retrotransposon, Tnt1. The maternal Tnt1 was transmitted to progenies, whereas the paternal Tnt1 was lost completely. Because TE mobilization can induce genome instability and gene disruption, the activities of TEs are under the control of host epigenetic mechanisms (Martienssen and Chandler 2013). Parisod et al. (2009) used a methylation-sensitive SSAP technique to show rapid epigenetic reorganization near retrotransposons in hybrid and allopolyploid Spartina genomes. Moreover, their study also revealed that genome alteration appeared preferentially in the maternal subgenome, and the environment of TEs was specifically affected by large maternal-specific methylation changes.
Retrotransposon-based insertional polymorphism (RBIP)
RBIP utilizes a primer designed on the basis of the LTR sequence and another primer from the genomic sequence near the LTR sequence (Paux et al. 2010). It detects polymorphism for the integration of an element at a particular locus to supply accurate DNA profiles. Retrotransposon insertions usually span several kilobase pairs. PCR amplification with a primer pair flanking the insertion sequences will produce an amplicon if the site is empty. However, if insertion occurred, the PCR amplification may yield a very long amplicon or not yield any amplicon because the insertion is too long to amplify. The incorporation of a primer within the inserted sequence will ensure amplification in the latter case. This three-primer system produces co-dominant RBIP markers, which provide extremely useful phylogenetic information because the retrotransposon insertions are irreversible. Using this three-primer system, Vitte et al. (2004) demonstrated that two distinct types of Asian rice varieties, Indica and Japonica, originated from two independent domestication events in Asia. RBIP is a very valuable resource for the protection of breeders’ rights, because it provides accurate, cultivar-specific DNA markers of insertion events that occur during cultivar development. In the hopes of developing markers for marker-assisted breeding in pears (Pyrus pyrifolia Nakai), Kim et al. (2012) generated 22 RBIP markers that are able to distinguish 61 of the 64 Japanese pear cultivars.
Class II DNA transposon-derived markers
Cut-and-paste transposition does not leave the original copy behind after transposition, so those transposons do not usually reach very high copy numbers in the genome, unlike the class I retrotransposons. Nevertheless, some class II DNA transposons (e.g., MITE, CACTA) are present in the genome with copy numbers from several thousands to hundreds of thousands (Bureau and Wessler 1992, 1994; Kunze and Weil 2002; Park and Kim 2012). This review introduces MITE and CACTA transposons as molecular markers for phylogenetic and genetic analyses using TD. TD is a technique that uses sequence-tagged site (STS) markers (Thomas and Scott 1993; Talbert et al. 1994; Sanchez et al. 1999). Korswagen et al. (1996) developed a TD protocol by modifying the STS technique to utilize a primer that is complementary to the internal TC-1 transposon and another primer that flanks the transposon. Van den Broeck et al. (1998) further refined the TD protocol to be utilized for multicopy elements. Figure 3 illustrates the CACTA-TD, which was employed in an analysis of an Arabidopsis thaliana population (Park et al. 2014).
The MITEs are an exceptional class II DNA transposon family. MITEs are small (< 500 bp) and carry no obvious ORFs to be mobilized themselves (Bureau and Wessler 1992, 1994; Feschotte et al. 2002a, b). However, they are present in several hundreds of thousands of copies in their host genomes, which distinguish them among the class II DNA transposons. The extremely abundant copies make the MITEs useful as molecular markers. Park et al. (2003a) isolated an MITE subfamily, Pangrangja, from some Gramineae species. MITE-TD using the terminal invert repeat (TIR) of the Pangrangja element revealed that the Pangrangja elements are common in species across the genus Oryza. Moreover, the Pangrangja MITE-TD profiles were matched with the geographical distribution of Oryza species in Asia, Africa, and Australia (Park et al. 2003b, c). Subsequent analysis of segregating populations showed that the Pangrangja MITEs are distributed evenly along all the chromosomes in rice (Kwon et al. 2006b) and maize (Lee et al. 2004). Because MITE sequences are short, amplification of the insertion polymorphism by PCR was also developed in rice. Monden et al. (2009) analyzed segregating lines of rice derived from a japonica × japonica cross by MITE insertion polymorphisms. They mined the mPing MITE from the rice genome database and designed a primer pair flanking the MITE loci. Of the 183 MITE loci analyzed, 150 loci showed polymorphisms between two japonica rice lines and were used to construct a recombinant genetic map for the analysis of quantitative trait loci. MITE-TD is also a good resource for detecting the transpositional activity of transposons. Jiang et al. (2003) used MITE-TD analysis to demonstrate that the transposition of mPing MITEs could be induced in tissue culture. mPing is a non-autonomous element and can be mobilized in the presence of the autonomous element Pong by genomic shocks including anther culture (Kikuchi et al. 2003). The new insertion bands were isolated from a gel and sequenced after TD gel display, which revealed that the mPing insertion mostly occurred in low-copy regions of the rice genome.
CACTA is a prototype class II DNA transposon superfamily that was discovered in maize by classical genetics as the Enhancer (En) (Peterson 1953) and Suppressor–Mutator (Spm) (McClintock 1954) elements. The TIRs of CACTA are short, ranging from 13 to 20 bp, and they have distinctive 5′-CACTA-3′ sequences in their termini (Kunze and Weil 2002). The CACTA superfamily is present in high copy numbers in plants and can be classified into several subfamilies. Each subfamily has sequence conservation of about 20–30 bp at their termini (Wicker et al. 2003). Kwon et al. (2005) utilized the consensus sequence in the TIR of the Rim2/Hippa CACTA subfamily for a CACTA-TD analysis in Oryza species. The phylogenetic dendrogram derived from the Rim 2/Hippa CACTA-TD was congruent with the geographic distribution of the A genome diploid Oryza species (Kwon et al. 2005, 2006a). The Rim 2/Hippa CACTA-TD profiles were highly reproducible and applicable to the construction of a high-density genetic map of Oryza sativa L. (Kwon et al. 2006b). CACTA elements are also highly abundant in Arabidopsis and Brassica species (Zhang and Wessler 2004). Park and Kim (2012) isolated all copies of the CACTA-like elements from the genome of A. thaliana and located them near the centromeric regions of all five chromosomes (Park and Kim 2012). Because the CACTA-TD profiles were revealed to be highly variable among different ecotypes of A. thaliana, the relationship between CACTA transposition and ecotype differentiation was analyzed by joint analyses of CACTA-TD and conventional AFLP among ecotypes. Although the transposition might not directly cause ecotype differentiation, the authors proposed that the insertion or excision of TEs into or out of critically functional genes could still be directly or indirectly involved in ecotype differentiation. In crop breeding, cultivar fingerprinting is an important technique for protecting breeders’ rights. CACTA-TD was also successfully applied in rapeseed. Lee et al. (2012) mined CACTA elements from the Brassica sequence database and designed a CACTA-TD experiment with 10 commercial rapeseed cultivars. The polymorphic CACTA-TD fragments were isolated from gels, and the sequences were determined to generate sequence-characterized amplified region (SCAR) markers, which led to the development of six CACTA-TD-derived transposon insertion SCAR (Ti-SCAR) markers for rapeseed.
Conclusion
Molecular markers are indispensable tools in modern and agricultural genetics (Schulman 2007; Henry 2013; Jiang 2013). Although several marker systems have been developed for plant genotypes, and abundantly available genome sequences promise to provide powerful molecular markers, none of the marker systems is a “jack-of-all-trades” for all concerns in plant genome analyses. RFLP, RAPD, and AFLP are based on sequence variation, and SSRP detects variation in the repeat numbers of simple sequence motifs. Transposon-based marker systems are based on the presence/absence of transposons at genetic loci. From the genomic projects for many eukaryotic organisms, it was realized that transposons are at the center of the dynamic evolution of many genomes. Next-generation sequencing techniques are generating sequence information at a speed never before seen (Varshney et al. 2009; Kelly and Leitch 2011; Edwards 2013). Moreover, computational biology allows us to mine genome-wide transposons and analyze their phylogenetic relationships (Xing et al. 2013). Thus, the marker systems that harness transposons are providing a new venue, in addition to the other marker systems using sequence variations or simple sequence motif variations, for the dissection of genomes in the genomic era.
References
Bachmann K (1994) Molecular markers in plant ecology. New Phytol 126:403–418
Bensen RJ, Johal GS, Crane VC, Tossberg JT, Schnable PS, Meely RB, Briggs SP (1995) Cloning and characterization of the maize An1 gene. Plant Cell 7:75–84
Bonchev G, Parisod C (2013) Transposable elements and microevolutionary changes in natural populations. Mol Ecol Res 13:765–775
Borevitz JO, Liang D, Plouffe D, Chang HS, Zhu T, Weigel D, Berry CC, Winzeler E, Chory J (2003) Large-scale identification of single-feature polymorphism in complex genome. Genome Res 13:513–523
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Branco CJ, Vieira EA, Malone G, Kopp MM, Malone E, Bernardes A, Mistura CC, Carvalho FIF, Oliveria CA (2007) IRAP and REMAP assessment of genetic similarity in rice. J Appl Genet 48:107–113
Bureau T, Wessler SR (1992) Tourist: a large family of inverted-repeat element frequently associated with maize genes. Plant Cell 4:1248–1283
Bureau T, Wessler SR (1994) Stowaway: a large-family of invert-repeat element family frequently associated with maize genes. Plant Cell 4:1283–1294
Chagné D, Gasic K, Crowhurst RN, Han Y, Bassett HC, Bowatte DR, Lawrence TJ, Rikkerink EHA, Gardiner SE, Korban SS (2008) Development of a set of SNP markers present in expressed genes of the apple. Genomics 92:353–358
Craig NL, Craigie R, Gellert M, Lambowitz AM (2002) Mobile DNA II. Am Soc Microbiol Press, Washington
Das L, Martienssen R (1995) Site-selected transposon mutagenesis at the hcf106 locus in maize. Plant Cell 7:287–294
Du C, Fefelova N, Caronna J, He L, Dooner HK (2009) The polychromatic Helitron landscape of the maize genome. Proc Natl Acad Sci USA 106:1191–1192
Edwards M (2013) Whole-genome sequencing for marker discovery. In: Henry RJ (ed) Molecular markers in plants. Wiley-Blackwell Pub, Ames, pp 21–34
Ellergren H (2004) Microsatellites: simple sequence with complex evolution. Nat Rev Genet 5:435–445
Fan F, Cui B, Zhang T, Ding G, Wen X (2014) LTR-retrotransposon activation, IRAP marker development and its potential in genetic diversity assessment of masson pine (Linus massoniana). Tree Genet Genomics 10:2013–2222
Fedoroff NV (2013) The discovery of transposition. In: Fedoroff NV (ed) Plant transposons and genome dynamics in evolution. Wiley-Blackwell Inc, Iowa, pp 3–14
Fedoroff NV, Bennetzen JL (2013) Transposon, genomic shock, and genome expansion. In: Fedoroff NV (ed) Plant transposons and genome dynamics in evolution. Wiley-Blackwell Inc, Iowa, pp 181–201
Feschotte C, Zhang X, Wessler SR (2002a) Miniature inverted-repeat transposable elements and their relationship to established DNA transposons. In: Craig NL, Cragie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. ASM Press, Washington, pp 1147–1158
Feschotte C, Jiang N, Wessler SR (2002b) Plant transposable elements: where genetics meets genomics. Nat Rev Genet 3:329–341
Finnegan DJ (1989) Eukaryotic transposable elements and genome evolution. Trends Genet 5:103–107
Ganal MW, Altman T, Röder MS (2009) SNP identification in crop plants. Curr Opin Plant Biol 12:211–217
Ganapathy PS, Scandalios JG (1973) Malate dehydrogenase isozymes in haploid and diploid Datura species. Their use as markers in somatic cell genetics. J Hered 64:186–188
Gore MA, Wright MH, Ersoz ES, Bouffard P, Szekeres ES, Jarvie TP, Hurwitz BL, Narechania A, Harkins TT, Grills GS et al (2009) Large-scale discovery of gene enriched SNPs. Plant Genome 2:121–133
Grzebelus D (2006) Transposon insertion polymorphism as a new source of molecular markers. J Fruit Ornam Plant Res 14:21–29
Henry RJ (2013) Evolution of DNA marker technology in plants. In: Henry RJ (ed) Molecular markers in plants. Wiley-Blackwell Pub, Ames, pp 3–20
Huang X, Lu G, Zhao Q, Liu X, Han B (2008) Genome-wide analysis of transposon insertion polymorphisms reveals intraspecific variation in cultivated rice. Plant Physiol 148:25–40
Hubby JL, Lewontin RC (1966) A molecular approach to the study of genic heterozygosity in natural populations, I. The number of alleles at different loci in Drosophila pseudoobscura. Genetics 54:577–594
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucl Acids Res 29(4):e25
Jiang GL (2013) Moleular markers and marker-assisted breeding in plants. In: Andersen SB (ed) Agricultural and biological science. Plant Breeding from Laboratories to Fields. doi:10.5772/52583
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR (2003) An active DNA transposon family in rice. Nature 421:163–167
Jing R, Vershinin A, Grzebyta J, Smýkal P, Marsshall D, Ambrose MJ, Ellis TN, Flavell AJ (2010) The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymoprphism (RIBP) marker analysis. BMC Evol Biol 10:44
Josefesson C, Dilkes B, Comai L (2006) Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol 16:1322–1328
Kalendar R, Schulman AH (2006) IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat Protocols 1:2478–2484
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman AH (1999) IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711
Kalendar R, Flavell AJ, Ellis T, Sjakste T, Moisy C, Schulman A (2011) Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 106:520–530
Kapitonov V, Jurka J (2001) Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA 98:8714–8719
Kashkush K, Feldman M, Levy AA (2002) Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160:1651–1659
Kelly LJ, Leitch IJ (2011) Exploring giant plant genomes with next-generation sequencing technology. Chromosome Res 19:939–953
Kikuchi K, Terauchi K, Wada M, Hirano HY (2003) The plant MITE mPing is mobilized in anther culture. Nature 421:167–170
Kim H, Terakami S, Nishitani C, Kurita K, Kanamori H, Katayose Y, Sawamura Y, Saito T, Yamamoto T (2012) Development of cultivar-specific DNA markers based on retrotransposon-based insertional polymorphism in Japanese pear. Breed Sci 62:53–62
Korswagen HC, Durbin RM, Smits MT, Plasterk RHA (1996) Transposon TC1-derived, sequence-tagged sites in Caenorhabiditis elegans as markers for genetic mapping. Proc Natl Acad Sci USA 93:14680–14685
Kumar A, Hirochika H (2001) Applications of retrotransposons as genetic tools in plant biology. Trends Plant Biol 6:127–134
Kumar R, Qiu J, Joshi T, Valliyodan B, Xu D, Nguyen HT (2007) Single feature polymorphism discovery in rice. PLoS One 2:e284
Kunze R, Weil CF (2002) hAT and CACTA superfamilies’ in plant genomes. ASM Press, Washington, pp 565–612
Kwon SJ, Park KC, Kim JH, Lee JK, Kim NS (2005) Rim 2/Hipa CACTA transposon display: a new genetic marker technique in Oryza species. BMC Genet 6:15
Kwon SJ, Lee JK, Hong SW, Park YJ, McNally KL, Kim NS (2006a) Genetic diversity and phylogenetic relationship in AA Oryza species as revealed by Rim 2/Hipa CACTA transposon display. Genes Gent Syst 81:93–101
Kwon SJ, Hong SW, Son JH, Lee JK, Cha YS, Eun MY, Kim NS (2006b) CACTA and MITE transposon distribution in a recombinant genetic map of rice. Mol Cells 21:360–366
Le QH, Bureau T (2004) Prediction and quality assessment of transposon insertion display data. Biotechniques 36:222–228
Lee SI, Kim NS (2014) Transposable elements and genome size variations in plants. Genomics Inform 12:87–97
Lee JK, Park JY, Choi SH, Kim JH, Choi JK, Min HK, Park CH, Kim NS (2004) Genetic mapping of maize with the intermated Mo17 × KW7 population using MITE-AFLP and SSR markers. Kor J Genet 26:63–72
Lee SI, Park KC, Ha MW, Kim KS, Jang YS, Kim NS (2012) CACTA transposon-derived Ti-SCARs for cultivar fingerprinting in rapeseed. Genes Genom 34:575–579
Leeton PR, Smyth DR (1993) An abundant LINE-like elements amplified in the genome of Lilium speciosum. Mol General Genet 237:97–104
Levin H (2002) Newly identified retrotranspoons of the Ty3/gypsy class in fungi, plants, and vertebrates. In: Craig NL, Cragie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. ASM Press, Washington, pp 684–701
Liu B, Wendel JF (2000) Retrotransposon activation followed by rapid repression in introgressed rice plants. Genome 43: 874–880. In: Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) Mobile elements II. American Society of Microbiology Press, Washington, DC
Lockton S, Ross-Ibarra J, Gaut BS (2008) Demography and weak selection drive patterns of transposable element diversity in natural Arabidopsis lyrata. Proc Nat Acad Sci USA 105:13965–13970
Mammadov J, Aggarwal T, Buyarapu R, Kumpatala S (2012) SNP markers and their impact on plant breeding. Int J Plant Genomics. doi:10.1155/2012/728398
Martienssen RA, Chandler VL (2013) Molecular mechanisms of transposon epigenetic regulation. In: Fedorof NV (ed) Transposons and genome dynamics in evolution. Wiley-Blackwell Pub, Ames, pp 71–92
McClintock B (1954) Mutations in maize and chromosomal abberrations in Neurospora. Carnegie Inst 53:254–260 Wanshingtonm Yearbook
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
Moisy C, Garrison KE, Meredith CP, Pelsy F (2008) Characterization of ten novel Ty1/copia-like retrotransposon families of the grapevine genome. BMC Genom 9:469
Monden Y, Naito K, Okumoto Y, Saito H, Oki N, Tsukiyama T, Ideta O, Nakazaki T, Wessler SR, Tanjisaka T (2009) High potential of a transposon mPing as a marker system in japonica × japonica cross in rice. DNA Res 16:131–140
Moran JV, Gilbert N (2002) Mammalian LINE-1 retrotransposons and related elements. In: Craig NL, Cragie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. ASM Press, Washington, pp 836–869
Oliver KR, Greene WK (2009) Transposable elements: powerful facilitators of evolution. BioEssays 31:703–714
Oliver KR, McComb JA, Greene W (2013) Transposable elements: powerful contributors to angiosperm evolution and diversity. Mol Biol Evol 5:1886–1901
Parisod C, Senerchia N (2012) Responses of transposable elements to polyploidy. In: Grandbastien M-A, Casacuberta J (eds) Plant transposable elements. Topics in current genetics, vol 24. Springer, Heidelberg, pp 147–168
Parisod C, Salmon A, Zerjal T, Tenaillon M, Grandbastien M-A, Ainouche M (2009) Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol 184:1003–1015
Park KC, Kim NS (2012) Differentiation of CACTA-like elements in Arabidopsis. In: Pontarotti P (ed) Evolutionary biology: mechanism and trend. Springer, Marseille, pp 325–342
Park KC, Chung CS, Song MT, Kim NS (2003a) A new MITE family, Pangrangja, in Gramineae species. Mol Cells 15:373–380
Park KC, Kim NH, Cho YS, Kang KH, Lee JK, Kim NS (2003b) Genetic variations of AA genome Oryza species measured by MITE-AFLP. Theor Appl Genet 107:203–209
Park KC, Lee JK, Kim NH, Shin YB, Lee JH, Kim NS (2003c) Genetic relationship and genetic variation in Oryza species using MITE-AFLP. Genes Genet System 78:235–243
Park Y-J, Lee JK, Kim N-S (2009) Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 14:4546–4569
Park KC, Park NI, Lee SI, Kim KS, Chang YS, Kim N-S (2014) A new active CACTA element and transposition activity in ecotype differentiation of Arabidopsis. Genes Genom 36:229–236
Paux E, Faure S, Choulet F, Roger D, Gauthier V, Martinant JP et al (2010) Insertion site-based polymorphism markers open new perspectives for genome saturation and marker assisted selection in wheat. Plant Biotech J 8:196–210
Peterson PA (1953) A multiple pale green locus in maize. Genetics 38:682–683
Petit M, Guidat C, Daniel J et al (2010) Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytol 186:135–137
Poczai P, Varga I, Laos M, Cseh A, Bell N, Valkonen JPT, Hyvönen J (2013) Advances in plant gene-targeted and functional markers: a review. Plant Methods 9:6
Prince JP, Tanksley SD (1992) Restriction fragment length polymorphism in plant breeding and genetics. Proceed R Soc Edinb Sect B Biol Sci 99:23–29
Rowold DJ, Herrara RJ (2000) Alu element and the human genome. Genetica 108:57–72
Sakamoto K, Okada N (1985) Rodent type 2 Alu family, rat identifier sequence, rabbit C family, and bovine or goat 73-bp repeat may have evolved from tRNA genes. J Mol Evol 22:134–140
Sanchez AC, Brar DS, Huang N, Li Z, Khush GS (1999) Sequence tagged site marker-assisted selection for three bacterial blight resistance genes in rice. Crop Sci 40:792–797
Schulman AH (2007) Molecular markers to assess genetic diversity. Euphytica 158:313–321
Schulman AH, Wicker T (2013) A field guide to transposable elements. In: Fedoroff NV (ed) Plant transposons and genome dynamics in evolution. Wiley Blackwell, Oxford, pp 15–40
Singer T, Fan Y, Chang HS, Zhu T, Hazen SP, Briggs SP (2006) A high resolution map of Arabidospis recombinant inbred lines by whole genome exon-array hybridization. PLoS Genet 2:e144
Syed NH, Flavell AJ (2007) Sequence specific amplification polymorphisms (SSAP): a multiple-locus approach for analyzing transposon insertions. Nat Protocols 1:2746–2752
Talbert LE, Blake NK, Chee PW, Blake TK, Magyar GM (1994) Evaluation of “sequence-tagged-site” PCR products as molecular markers in wheat. Theor Appl Genet 87:789–794
Tam SM, Mhiri C, Vogelaar A, Kerkveld M, Pearce SR, Grandbastien MA (2005) Comparative analyses of genetic diversities within tomato and pepper collections detected by retrotransposon-based SSAP, AFLP, and SSR. Theor Appl Genet 110:819–831
Tanksley SD (1983) Molecular markers in plant breeding. Plant Mol Biol Rep 1:3–8
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. Nat Biotechnol 7:257–264
Tauts D, Renz M (1984) Simple sequence are ubiquitous repetitive components of eukaryotic genomes. Nucl Acids Res 12:4127–4138
Temnykh S, DeClerk G, Lukashova A, Lipovich L, Cartihour S, McCouch S (2001) Computational and experimental analysis microsatellite in rice (Oryza sativa L.): frequency, length variation, transposon association, and genetic marker potential. Genome Res 11:1441–1452
Thomas MR, Scott NS (1993) Microsatellite repeats in grapevine reveal DNA polymorphism when analyzed as sequence-tagged sites (STSs). Theor Appl Genet 86:985–990
Van den Broeck D, Maes T, Sauer M, Zethof J, De Keukelerie P, D’hauw M, Van Montagu M, Gerats T (1998) Transposon display identifies individual transposable elements in high copy number lines. Plant J 13:121–129
Varshney RK, Nayak SN, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–529
Vitte C, Ishii T, Lamy F, Brar D, Panaud O (2004) Genomic paleontology provides evidence for two distinct origins of Asian rice (Oryza sativa L.). Mol Gen Genomics 272:504–511
Vos P, Hogers R, Bleeker M, Reijans M, van der Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucl Acids Res 21:4407–4414
Voytas DF, Boeke JD (2002) Ty1 and Ty5 of Saccaromyces cereviceae. In: Craig NL, Cragie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. ASM Press, Washington, pp 631–662
Vukich M, Schulman AH, Giordani T, Natali I, Kalendar R, Cavallini (2009) Genetic variability in sunflower (Helianthus annus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor Appl Genet 11:1027–1038
Wicker T, Guyot R, Yahiaouri M, Keller B (2003) CACTA transposons in Triticeae. A diverse family of high-copy repetitive elements. Plant Physiol 132:52–63
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Willams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acids Res 18:6531–6535
Woodrow P, Pontecorvo G, Fantaccione S, Fuggi A, Kafantaris I, Parisi D, Carillo P (2010) Polymorphism of a new Ty1-copia retrotransposon in durum wheat under salt and light stresses. Theor Appl Genet 121:311–322
Xing J, Witherspoon DJ, Jorde LB (2013) Mobile element biology: new possibilities with high-throughput sequencing. Trends Genet 29:280–289
Zhang X, Wessler SR (2004) Genome-wide comparative analysis of the transposable elements in the related species Arabidopsis thaliana and Brassica oleracea. Proc Natl Acad Sci USA 101:5589–5594
Zhuang J, Wang J, Theurkarf W, Weng Z (2014) TEMP: a computational method for analyzing transposable element polymorphism in populations. Nucl Acids Res. doi:10.1093/nar/gku323
Acknowledgments
This work was funded by a grant from Golden Seed Project, Ministry of Agriculture, Food and Rural Affairs (MAFRA), Ministry of Oceans and Fisheries (MOF), Rural Development of Korea (RDA), and Korea Forest Service (KFS).
Conflict of interest
Authors declare no conflict of interest on the contents of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Roy, N.S., Choi, JY., Lee, SI. et al. Marker utility of transposable elements for plant genetics, breeding, and ecology: a review. Genes Genom 37, 141–151 (2015). https://doi.org/10.1007/s13258-014-0252-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-014-0252-3