
Abstract
Common bean is the principal grain legume of North-India including Jammu & Kashmir and Ladhak. Owing to its high protein content, abundance of various micronutrients and essential amino acids and low cost, it is sometimes referred to as poor man’s meat. It is a very diverse crop and presents a rich reservoir of genetic resource to be explored. In the present study, we collected 102 common bean genotypes from different north western Himalayan regions of Jammu & Kashmir and Ladakh. This germplasm was then systematically purified and then characterised using SSRs. In order to examine the genetic diversity, various parameters were considered. The PIC values for the SSR loci ranged from 0.738 to 0.966 with an average of 0.899. The gene diversity between common bean genotypes ranged from 0.751 to 0.967. The major allele frequency ranged from 0.076 to 0.469 and the expected heterozygosity ranged from 0 to 0.135. Further, model based STRUCTURE analysis generated three subpopulations that correspond to distance based groups. Furthermore, the PCR assay for phaseolin locus led to the characterization of 81 genotypes into Meso-American and Andean types. Out of 81 genotypes, 40 possessed “S” type phaseolin and 41 possess “T” type phaseolin. The results of this study revealed the origin of common bean landraces grown in the north western regions of Jammu & Kashmir and Ladakh, India. The characterised material can be planned for conservation and can further be used as a genetic resource for improvement of common bean.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Common bean (Phaseolus vulgaris L. 2n = 2x = 22) is a herbaceous annual plant grown worldwide for its edible bean, popularly consumed both dry and green bean. Being an important component of sustainable agriculture, it is regarded as the ‘Grain of Hope’ [39, 56]. Genetic analysis of common bean shows that it is originated from Mesoamerica (Mexico, Central America, and Colombia) and presumably Andes (southern Peru, Bolivia, and Argentina) [20]. Six races of common bean have been identified within these gene pools which include three Andean (Chile, Peru and Nueva Granada) races and three Mesoamerican (Durango, Mesoamerican and Jalisco) races, all of these have been distinguished biochemically and morphologically [52, 53]. In addition to these six races, Guatemala race has been identified in Central America which includes climbing beans [3]. All these races have been analysed using various molecular markers including Isozymes, RAPDs and SSRs to obtain insights about their genetic diversity. PCR based simple sequence repeats (SSRs) are short segments of DNA repeats (2–6 nucleotide long) found multiple times in the genome. Among all the markers, SSRs have been used for genetic diversity and population structure studies from time to time in several crop plants and their related wild species e.g., rice [50]; Chinese bread wheat [38]; common bean [11, 33, 34, 58]; Wild Rhubarb [14]; Kewda [40]. Due to the fast rate of mutations and variability [38, 47] in common bean, SSRs are suitable for genetic mapping and evaluating the species diversity [31, 35, 37].
Also, SSR markers are highly polymorphic and multi allelic that can amplify up to 25 alleles per locus and are abundant and widely distributed in both gene coding and non-coding regions of higher eukaryotes. Gaitán-Solís et al. [16] first time developed SSR markers for common beans to evaluate genetic diversity of cultivated and wild species. SSR based genetic maps have been constructed in common beans to evaluate the intraspecific genetic diversity [5, 16, 57]. SSRs have been divided into genic and genomics SSRs. Genic SSRs/microsatellites are those that are found within or closely associated with gene sequences in the genome whereas, genomic SSRs are associated with non-gene containing genomics regions [5, 54]. The genic SSR are well conserved and comprise diverse types of sequences of SSRs in the genome and have been frequently used for diversity analysis [5].
Phaseolin gene based characterisation of common bean germplasm has been done to understand the existence of gene pools in the collected material. The Phaseolin is major protein in common beans coded by 6–10 small family of genes which are tightly linked in the D7 linkage group [10, 21, 44]. Multiple domestication and reduction in the genetic diversity of common beans was found by studying the genetic variation patterns in Phaseolin protein locus by polyacrylamide gel electrophoretic studies [17, 19, 30]. Two types of Phaseolin proteins have been identified that include S and T types [10, 19]. High levels of sequence similarity was found between S and T phaseolin types after subjecting the genomes of common beans to DNA sequence analysis [2, 29]. It has been found that T phaseolin gene comprises of α and β sub-families. The members of α sub-family consists of tandem repeats sequences of 15 bp in 4th exon and 27 bp in 6th exon, whereas, these repeats are not found in β sub-families of T Phaseolin [29]. Cultivated crops are well characterized both morphologically and through molecular analysis whereas as wild crops need to be further investigated by assessing difference in morphological characters, protein (Phaseolin) type and by using molecular markers [5]. In 2014, the genome of common bean became available, and that made it possible to compare and understand the similarities and differences which make these species unique and important for human nutrition around the globe [25]. The in-depth knowledge of diversity of a specific crop is prerequisite for setting up an effective breeding program. This goal can be achieved, by making use of available diverse landraces that are actual repositories of novel genes for evolution of quality traits. The information of association mapping, mining of alleles for novel genes can be possible by evaluating population structure and genetic diversity of a specific crop [54]. In the present study, we employed SSR markers to elucidate the genetic diversity and population structure of Phaseolus vulgaris genotypes collected from foothills of the Himalayan region of Jammu and Kashmir and Ladakh, in an attempt to understand the variability among them. Moreover, Phaseolin gene based diversity was studied to discrimate the collected material based on their respective gene pool.
Materials and methods
Collection of plant material
The genetic material in the present studies included seeds of 102 genotypes collected from North-West Himalyan regions of Jammu and Kashmir and Ladhak, India (Supplementary table 1). The samples were collected from the local farmers and then subjected to field trials in the experimental fields at SKUAST-K, Shalimar campus (34.1485° N, 74.8696° E) to purify the material and were then subjected to analysis.
Genomic DNA isolation, SSR genotyping and scoring of bands
Fresh leaves from the 2 week old seedlings grown in cups were harvested for genomic DNA isolation. The genomic DNA was isolated by following Doyle and Doyle [12] method. The isolated DNA was purified and dissolved in 1X TE Buffer and stored at 4 °C. Quantification of DNA was done by running samples on 0.8% Agarose gel stained with ethidium bromide along with known standards and the results were visualized in Gel Documentations System (Syngene, Genius).
To carry out molecular characterization a set of 11 SSRs (detailed in supplementary Table 2) were used. One SSR of each linakge group was selected. The PCR amplification was carried for 5 μl reaction mixture containing 50 ng DNA template, 2.5 μl of 2X KAPA Taq ReadyMix (Cat. No. KK1024) manufactured by Kapa Biosystems (Sigma-aldrich.com) 0.37 μl of 0.74 pM of each primer (Forward and Reverse) and 1.06 μl of sterile water. The amplification reaction was carried out in a gradient master cycler (Applied Biosystems, Thermo Scientific). An initial denaturation step for 4 min, followed by a loop of 35 cycles each consisting of denaturation (at 94 °C for 30 s), annealing (at 47–55 °C for 30 s) and extension (at 72 °C for 30 s) was programmed. The final extension was performed at 72 °C for 7 min. The amplified products were resolved on 3% agarose gel subjected to 125 V for 1 h the poorly resolved SSRs were further resolved on silver stained PAGE (poly acrylamide gel electrophoresis). The gels were visualized in gel documentation system with inbuilt software ALPHA SA system for scoring of bands. The bands found within the range of expected base pair (bp) were scored in allelic form according to their fragment size (bp) corresponding to the 100 bp molecular weight marker (Invitrogen; Cat. No. 10488-053).
Data analysis based on SSR markers
Eleven SSRs were employed to study various discriminatory parameters among 102 genotypes of common bean by using Power Marker [32] and GenALEx 6.51 [45]. The unweighted neighbor joining tree was constructed by analyzing dissimilarity matrix using shared allele index in DARwin software [46]. Nei’s coefficient with bootstrap protocol of resampling across markers and individuals from allele frequencies was employed to construct the genetic distance between accessions [41]. Principal coordinate analysis (PCoA) was also performed by DARwin software [46]. Population structure of the genotypes under investigation was obtained by STRUCTRE version 2.3.4 software [48] by setting programme at 50,000 of each burn-in and MCMCs with 10 replications of each K (1–10). The results obtained by STRUCTURE software were further analyzed in STRUCTURE HARVESTER for finding out the best K value [13, 15]. In addition, the STRUCTURE software was used to estimate the level of genetic differentiation or wright fixation of F statistics (FST) simultaneously [55]. Further analysis of molecular variance (AMOVA) was performed by GenALEx 6.51 [45].
Phaseolin marker analysis
To classify the common bean genotypes based on their gene pools (Meso American and Andean), phaseolin locus was amplified on 81 genotypes of common bean. The primer for phaseolin gene with sequence for forward primer 5′-AGCATATTCTAGAGGCCTCC-3′, and the primer sequence for the reverse primer 5′-GCTCAGTTCCTCAATCTGTTC-3′ were selected from Kami et al. [29]. The procedure adopted for the amplification of phaseolin locus, was similar as adopted for SSR genotyping, which is detailed above. The scoring of phaseolin marker was done as per Kami et al. [29].
Results
SSRs based genotyping
A total of 102 genotypes were amplified by employing 11 SSRs. An average of 30 alleles per SSRs were amplified among 102 common bean genotypes with maximum 47 alleles in BM184 and minimum 13 alleles in BM98 (Table 1). The major allelic frequency varied from 0.076 (BM-210) to 0.469 (BMC-234) with an average mean of 0.216 (Table 1). The gene diversity (expected heterozygosity) values ranged from 0.751 (BMC-234) to 0.967 (BM-210) with an average of 0.904. The mean Polymorphic Information Content (PIC) value was found to be 0.899 with minimum value of 0.738 (BMC-234) and maximum of 0.952 (BM-210) (Table 1). Further, 11 SSRs revealed more than 50% population with no heterozygosity. Mean value for inbreeding coefficient within each individual was found to be 0.985 raging from 0.900 to 1.00 (Table 1). Moreover, average Fixation index per SSR was 0.094 with minimum value of 0.037 to maximum value of 0.256 (Table 1).
Genetic diversity and population structure analysis
The dendrogram illustrates the relationship among 102 genotypes of common bean. The genotypes were classified into three major cluster based on the genetic variation (Fig. 1a). Cluster I is sub-divided into two sub-clusters that includes 34 genotypes and cluster II is sub-divided into sub-clusters that includes 25 genotypes whereas, the cluster III sub-divided into other sub-clusters that includes 43 genotypes as detailed in Supplementary table 1. The PCoA also divided the genotypes into three groups (Fig. 1b).

a Dendrogram illustrating genetic relationship among 102 genotypes using 11 SSR primers, b principal coordinate analysis of 102 genotypes constructed by DARwin software
STRUCTURE analysis revealed formation of three populations with slight mixing of genotypes as represented in population structure plot (Fig. 2b). The assumed values of probable sub-populations (K) were ascertained by choosing higher ΔK value, with respect to the number of clusters inferred by STRUCTURE software (Fig. 2a). The individuals were assigned to sub population based on membership probability ≥ 80%. Subpopulation I consisted of 30 (29.4%), subpopulation II consisted of 49 (48%), subpopulation III consisted of 22 (21.5%) genotypes with only 1 genotype as admixture. Genetic differentiation between the three sub-populations ranging from 0.09 to 0.17 indicating that all the three population groups were significantly different from each other (Table 2). Further the expected heterozygosity ranged from 0.84 to 0.88 and FST values from 0.08 to 0.14 for three subpopulations (Table 3).

a Graphical representation of the optimal number of groups in the program STRUCTURE inferred using the criterion of Evano et al. [15]. b STRUCTURE plot of membership coefficients for all the accessions of common bean in the study sample sorted in the same order and classified according to successive selected preset K values ranging from 1 to 10. For K = 3 the groups are identified

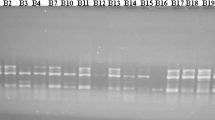
Results from phaseolin PCR assay. 8% polyacrylamide gel M depicts ladder in 1st and last well, genotype 1–51 from 2nd to second last well. T and S depicts T and S type of phaseolin; and-depicts no amplification
Analysis of molecular variance
Three common bean populations generated from structural analysis were also subjected to analysis of molecular variance (AMOVA) to estimate the percentage of variation among populations, among individuals and within individuals. Out of the total genetic variance among populations, 8% was attributed to the populations based on structure, 90% was attributed among individuals whereas 2% difference was attributed to within the individuals (Table 4).
Phaseolin gene based classification of germplasm
Amplification of phaseolin locus based on presence of either “S” or “T” type phaseolin was observed in 81 common bean genotypes. As such these 81 genotypes were classified into Mesoamerican or Andean origin. 40 genotypes (39.22%) with “S” type band were classified to be having Mesoamerican origin where as the 41 genotypes (40.20%) with “T” type band were classified to be having Andean origin (Fig. 3).
Discussion
Common bean is a widely cultivated crop in the Himalayan regions of Jammu and Kashmir. For the nutritional improvement of crop plants to combat the food and nutritional insecurity, knowledge of genetic diversity is crucial. The accomplishment of well-designed breeding programs needs germplasm with a high level of genetic diversity. So evaluation of genotypes for diversity analysis is important for its utilization in different breeding programs [39]. Majorly two strategies have been employed to assess the genetic makeup of crop plants that is morphological and molecular marker analysis. Morphological markers are easily affected by environmental and other factors, making it a very inaccurate method to characterize crop plants. Molecular markers like SSRs on the other hand, are accurate and reliable tools to assess the genetic variability and have been frequently used in common beans and other legumes [7, 18, 22, 24–27, 49].
In current study, SSR markers were successfully used to assess the genetic diversity among 102 common bean genotypes collected from Himalayan regions of Jammu and Kashmir and Ladakh. These SSRs were chosen from an abundance of markers available in public domain and were selected such that they cover all the linkage groups [5, 42]. In the present study, we observed an average number of 30 alleles per SSR. The results were nearly similar to Blair et al. [6]. The reason for high number of alleles per SSR might be the use of genomic SSR in this study, as genomic SSRs can resolve within gene pool variation [6]. SSR based polymorphism information content (PIC) can be employed for screening appropriate markers to construct the genetic maps, association mapping and carrying phylogenetic analysis [1]. The PIC values reveal the quality of the marker and its capability to detect the genetic variability based on preliminary studies [9]. Biallelic nature of dominant and co-dominant markers like ISSR and SSR respectively results in very low PIC values [48]. In present studies the values of PIC ranged from 0.738 to 0.952, the average value remained to 0.899. The high level of polymorphism is due to huge diversity among genotypes and selection of highly polymorphic markers based on earlier studies. Metais et al. [36] found PIC ranging from 0.05 to 0.83 after subjecting 20 genotypes of common beans to SSR marker analysis. In addition, Gomez et al. [23] assessed 60 genotypes of common beans to SSR markers analysis and found PIC values ranging from 0.03 to 0.70, suggesting that PIC values help to elucidate the complexity of diversity depending on number and genotype diversity. It has been observed that lower PIC values are obtained from closely related genotypes and higher values for genetically distant genotypes. Other frequently used parameter for assessing the genetic variability is gene diversity and heterozygosity. The values of PIC and gene diversity were found to be nearly similar which could be due to the large number of alleles per SSR. Variation in gene diversity and heterozygosity has been observed in earlier common bean diversity studies [23, 36, 42, 43, 51]. Use of germplasm from different geographical locations, availability of different marker systems and scoring pattern might lead to difference in these parameters. The structure of genetic variation within and among the population is greatly influenced by the Wright’s F-statistics (FIS, FST) [28]. FST is directly linked to the variance in allele frequency among populations whereas inversely to the degree of resemblance among individuals within populations. In our study, FST over all loci across common bean germplasm is 0.094, indicating a low degree of genetic differentiation among subpopulations. The estimates of within-subpopulation inbreeding coefficients (FIS) were considerably higher in our study (Table 1). High FIS implies a considerable degree of inbreeding [28].
Dividing the population based on the geographical location is an important parameter for studying evolution of a particular species. The unweighted neighbor joining tree constructed by DARWin software divided the collected germplasm into three clusters which distributed the genotypes based on the place of collection. Further the PCoA also divided the germplasm into three clusters. Moreover, population structure analysis is based on bayesian method and distributes the individual of a population based on ΔK. In our study, population structure analyses divided the genotypes into three populations (K = 3). As such, in the present study the germplasm was classified into three groups, based on cluster analysis, PCoA, structure analysis that has slight differences due to use of different algorithms. Different and large number of molecular markers and germplasm might result in variation in the population structure of that species. Partial reproductive isolation and lower genetic drift might also have attributed to the variation in diversity and population structure analysis. In case of common bean population structure analysis earlier studies have divided the common bean population into 2–6 sub population [8, 32, 42, 43, 51, 58]. Microsatellites have been previously used for the analysis of diversity in common bean breeding lines from Canada [57] in wild accessions and related species [15], in snap beans [36] and in dry bean land-races from Europe and Nicaragua [23]. Further, AMOVA analysis was also found to be in accordance with other results.
Common bean has been divided into two major ecogeographical gene pools i.e. Mesoamerica and the Andes 7000–8000 years ago. However, with the evolution process, common bean from Ecuador and northern Peru formed an intermediate (I) type gene pool between earlier two [29]. This intermediate type of gene pool was also confirmed by the phaseolin marker study in common bean germplasm [29]. These gene pools are characterized by partial reproductive isolation and could be seen in both wild and domesticated common bean genotypes [20, 30]. Origin and domestication of common bean germplasm can be known by Phaseolin marker. This marker helps in classification of common bean germplasm based on its origin [19, 29]. The presence of S type allele in the genotype shows its origin from Andean gene pool where as the presence of T type allele in the genotype shows its origin from Mesoamerican gene pool. In our study, we were able to characterize the common bean germplasm collected from Himalayan region into two gene pools Mesoamerican and Andean. Moreover, we could not find any genotype with I type gene pool, the reason might be relict population of I type as it represents only a small fraction of genetic diversity of the ancestral population [4].
Conclusion
The use of SSR markers for assessing genetic diversity and population structure of common beans from different north western Himalayan regions have shown significant levels of genetic variation that will serve as an important genetic resource. The study further unraveled the gene pool of 81 genotypes collected in the present investigation. The insights provided by this study will serve as a foothold in formulating strategies for conservation of these landraces. The identification of gene pool of each landrace will help the breeders to understand their evolution. This will also help in designing crossing programmes between and among genotypes of same or different gene pools for developing various mapping population for QTL/marker gene identifications which in turn can lead to the development of improved common bean varieties.
References
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME. Optimizing parental selection for genetic linkage maps. Genome. 1993;36:181–6.
Anthony JL, Vonder Haar RA, Hall TC. Nucleotide sequence of alpha phaseolin gene from Phaseolus vulgaris. Nucleic Acids Res. 1990;18:3396.
Beebe SE, Skroch PW, Tohme J, Duque MC, Pedraza F, Nienhuis J. Structure of genetic diversity among common bean landraces of middle American origin based on correspondence analysis of RAPD. Crop Sci. 2000;40:264–73.
Bitocchi E, Nanni L, Bellucci E, Rossi M, Giardini A, Zeuli PS, Logozzo G, Stougaard J, McClean P, Attene G, Papa R. Mesoamerican origin of the common bean (Phaseolus vulgaris L.) is revealed by sequence data. Proc Natl Acad Sci. 2012;109(14):E788–96.
Blair MW, Pedraza F, Buendia HF, Gaitán-Solís E, Beebe SE, Gepts P, Tohme J. Development of a genome-wide anchored microsatellite map for common bean (Phaseolus vulgaris L.). Theor Appl Genet. 2003;107:1362–74.
Blair MW, Giraldo MC, Buendia HF, Tovar E, Duque MC, Beebe SE. Microsatellite marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet. 2006;113(1):100–9.
Blair MW, Torres MM, Pedraza F, Giraldo MC, Buenda HF, Hurtado N. Development of microsatellite markers for common bean (Phaseolus vulgaris L.) based on screening of non-enriched, small-insert genomic libraries. Genome. 2009;52:772–82.
Blair MW, Soler A, Cortés AJ. Diversification and population structure in common beans (Phaseolus vulgaris L.). PLoS ONE. 2012;7(11):e49488. https://doi.org/10.1371/journal.pone.0049488.
Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314.
Brown JWS, McFerson JR, Bliss FA, Hall TC. Genetic divergence among commercial classes of Phaseolus vulgaris in relation to phaseolin pattern. Hort Sci. 1982;17:752–4.
Cortés AJ, Chavarro MC, Blair MW. SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet. 2011;123:827–45.
Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12(13):39–41.
Earl D, von Holdt B. STRUCTURE harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Ekincialp A, Erdinc C, Turan S, Cakmakci O, Nadeem MA, Baloch FS, Sensoy S. Genetic characterization of Rheum ribes L. (wild rhubarb) genotypes in lake van basin of Turkey through ISSR and SSR markers. Int J Agric Biol. 2019;21:795–802.
Evano G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Gaitan-Solis E, Duque MC, Edwards K, Tohme J. Microsatellite repeats in common bean (Phaseolus vulgaris L.): isolation, characterization, and cross-species amplification in Phaseolus spp. Crop Sci. 2002;42:2128–36.
Gepts P. Biochemical evidence bearing on the domestication of Phaseolus (Fabaceae) beans. Econ Bot. 1990;44:28–38. https://doi.org/10.1007/BF02860473.
Gepts P. The use of molecular and biochemical markers in crop evolution studies. Evol Biol. 1993;27:51–94.
Gepts P, Bliss FA. Phaseolin variability among wild and cultivated common beans (Phaseolus vulgaris) from Colombia. Econ Bot. 1986;40:469–78. https://doi.org/10.1007/BF02859660.
Gepts P, Debouck D. Origin, domestication, and evolution of the common bean (Phaseolus vulgaris L.). In: Schoonhoven AV, editor. Common beans: research for crop improvement. Cali: CIAT; 1991. p. 7–53.
Gepts P, Nodari R, Tsai R, Koinange EMK, Llaca V, Gilbertson R, Guzman P. Linkage mapping in common bean. Annu Rep Bean Improv Coop. 1993;36:xxiv–xxxviii.
Gepts P, Osborn TC, Rashka K, Bliss FA. Phaseolin-protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris): evidence for multiple centers of domestication. Econ Bot. 1986;40:451–68.
Gomez E, Ferreras L, Toresani S. Soil bacterial functional diversity as influenced by organic amendment application. Biores Technol. 2006;97(13):1484–9.
Guerrero Castro NA, Isidra-Arellano MC, Mendoza-Cozatl DG, ValdésLópez O. Common bean: a legume model on the rise for unravelling responses and Intraspecific diversity in Phaseolus vulgaris. Genome. 2016;37(5):751–7.
Gyang PJ, Nyaboga EN, Muge EK. Molecular characterization of common bean (Phaseolus vulgaris L.) genotypes using microsatellite markers. J Adv Biol Biotechnol. 2017;13(2):1–15.
Hanai LR, Santini L, Camargo LEA, Fungaro MHP, Gepts P, Tsai SM, Vieira MLC. Extension of the core map of common bean with EST-SSR, RGA, AFLP and putative functional markers. Mol Breed. 2010;25(1):25–45.
Hao C, Wang L, Ge H, Dong Y, Zhang X. Genetic diversity and linkage disequilibrium in Chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS One. 2011;6(2):e17279. https://doi.org/10.1371/journal.pone.0017279.
Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet. 2009;10:639–50.
Kami J, Velasquez VB, Debouck DG, Gepts P. Identification of presumed ancestral DNA sequences of phaseolin in Phaseolus vulgaris. Proc Natl Acad Sci. 1995;92(4):1101–4. https://doi.org/10.1073/pnas.92.4.1101.
Koenig R, Singh SP, Gepts P. Novel phaseolin types in wild and cultivated common bean (Phaseolus vulgaris, Fabaceae). Econ Bot. 1990;44:50–60.
Lee M. DNA markers and plant breeding programs. Adv Agron. 1995;55:265–344.
Liu K, Muse SV. Power marker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–9.
Mahajan R, Zargar SM, Salgotra RK, Singh R, Wani AA, Nazir M, Sofi PA. Linkage disequilibrium based association mapping of micronutrients in common bean (Phaseolus vulgaris L.): a collection of Jammu & Kashmir, India. 3 Biotech. 2017;7:295. https://doi.org/10.1007/s13205-017-0928-x.
Mahajan R, Zargar SM, Singh R, Salgotra RK, Farhat S, Sonah H. Population structure analysis and selection of core set among common bean genotypes from Jammu and Kashmir, India. Appl Biochem Biotechnol. 2017;182(1):16–28. https://doi.org/10.1007/s12010-016-2307-1.
Matus IA, Hayes PM. Genetic diversity in three groups of barley germplasm assessed by simple sequence repeats. Genome. 2002;45:1095–106.
Métais I, Hamon B, Jalouzot R, Peltier D. Structure and level of genetic diversity in various bean types evidenced with microsatellite markers isolated from a genomic enriched library. Theor Appl Genet. 2002;104(8):1346–52.
Mitchell SE, Kresovich S, Jester CA, Hernandez CJ, Szewc-McFadden AK. Application of multiplex-PCR and fluorescence based, semi-automated allele sizing technology for genotyping plant genetic resources. Crop Sci. 1997;37:617–24.
Morgante M, Olivieri AM. PCR-amplification of microsatellites as markers in plant genetics. Plant J. 1993;3:175–82.
Nadeem MA, Karaköy T, Yeken MZ, Habyarimana E, Hatipoğlu R, Çiftçi V, Nawaz MA, Sönmez F, Shahid MQ, Yang SH, Chung G, Baloch FS. Phenotypic characterization of 183 Turkish common bean accessions for agronomic, trading, and consumer-preferred plant characteristics for breeding purposes. Agronomy. 2020;10:272.
Nasim N, Sandeep IS, Sahoo A, Das S, Panda MK, Acharya L, RamaRao VV, Nayak S, Mohanty S. Population genetic structure and diversity analysis in economically important Pandanus odorifer (Forssk.) Kuntze accessions employing ISSR and SSR markers. Ind Crops Prod. 2020;143:111894. https://doi.org/10.1016/j.indcrop.2019.111894.
Nei M. Genetic distance between populations. Am Nat. 1972;106:283–92.
Nemli S, Asciogul TK, Kay HB, Kahraman A, Siyok DE, Tanyolac B. Association mapping for five agronomic traits in the common bean (Phaseolus vulgaris L.). J Sci Food Agric. 2014. https://doi.org/10.1002/jsfa.6664.
Okii D, Tukamuhabwa P, Kami J, Namayanja A, Paparu P, Ugen M, Gepts P. The genetic diversity and population structure of common bean (Phaseolus vulgaris L.) germplasm in Uganda. Afr J Biotechnol. 2014;13(29):2935–49.
Osborne TB. The vegetable proteins. London: Longmans and Green; 1907.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28:2537–9.
Perrier X, Jacquemoud-Collet JP. DARwin software. 2006. http://darwin.cirad.fr/darwin. Accessed 20 June 2019
Powell W, Machray GC, Provan J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996;1:215–22.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Roldàn-Ruiz I, Dendauw J, Van Bockstaele E, Depicker A. AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp.). Mol Breed. 2000;6:125–34.
Salgotra RK, Gupta BB, Bhat JA, Sharma S. Genetic diversity and population structure of basmati rice (Oryza sativa L.) germplasm collected from North Western Himalayas using trait linked SSR markers. PLoS One. 2015;10(7):e0131858. https://doi.org/10.1371/journal.pone.0131858.
Sicard, Hanai LR, De Campos T, Camargo LEA, Benchimol LL, De Souza AP, Melotto M, Siqueira MVBM. Development, characterization, and comparative analysis of polymorphism at common bean SSR loci isolated from genic and genomic sources. Genome. 2007;50(3):266–77.
Singh SP, Gepts P, Debouck D. Races of common bean (Phaseolus vulgaris, Fabaceae). Econ Bot. 1991;45:379–96.
Singh SP, Nodari R, Gepts P. Genetic diversity in cultivated common bean: I. Allozymes. Crop Sci. 1991;31:19–23.
Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48–55.
Wright S. The genetic structure of populations. Ann Eugenics. 1951;15:323–54.
Yeken MZ, Nadeem MA, Karaköy T, Baloch FS, Çiftçi V. Determination of Turkish common bean germplasm for morpho-agronomic and mineral variations for breeding perspectives in Turkey. KSU J Agric Nat. 2019;22(Suppl: 1):38–50. https://doi.org/10.18016/ksutarimdoga.vi.549996.
Yu K, Park SJ, Poysa V, Gepts P. Integration of simple sequence repeat (SSR) markers into a molecular linkage map of common bean (Phaseolus vulgaris L.). J Hered. 2000;91:429–34.
Zargar SM, Farhat S, Mahajan R, Bhakhri A, Sharma A. Unravelling the efficiency of RAPD and SSR markers in diversity analysis and population structure estimation in common bean. Saudi J Biol Sci. 2016;23(1):139–49.
Acknowledgements
SMZ is grateful to NMHS GBPNIHESD, Almora, Uttrakhand, India for financial support of this work (Project Sanction Order No. GBPNI/NMHS17-18/SG24/622).
Author information
Authors and Affiliations
Contributions
HB have contributed in conducting the laboratory experiment. ZB was involved in the collection of common bean landraces and further purified the lines. RM did the analysis and helped in preparing the manuscript and in revision of manuscript. MN was involved in collection and data analysis. RAM helped in preparing the manuscript. FAN guided in the field related work and checked the manuscript. SMZ has designed the experiment, helped in analysis, prepared the final manuscript, assisted in revision of manuscript and got grant for undertaking this research work.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Corresponding Editor: Manoj Prasad.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bashir, H., Bashir, Z., Mahajan, R. et al. Molecular characterization and insights into the origin of common bean (Phaseolus vulgaris L.) landraces of north western Himalayas. Nucleus 63, 271–279 (2020). https://doi.org/10.1007/s13237-020-00323-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13237-020-00323-2