
Abstract
Anthracnose disease caused by the Colletotrichum gloeosporioides species complex is a major problem worldwide. In this study, we investigated the phylogenetic diversity of 207 Indian Colletotrichum isolates, associated with symptomatic and asymptomatic tissues of mango, belonging to this species complex. Phylogenetic analyses were performed based on a 6-gene dataset (act, cal, chs1, gapdh, ITS and tub2), followed by ApMat sequence-analysis. The ApMat-based phylogeny was found to be superior as it provided finer resolution in most of the species-level clades. Importantly, the ApMat marker identified seven lineages within C. siamense sensu lato, including C. jasmini-sambac, C. hymenocallidis, C. melanocaulon, C. siamense sensu stricto and three undesignated, potentially novel lineages. In this study, C. fragariae sensu stricto, C. fructicola, C. jasmini-sambac, C. melanocaulon and five undesignated, potentially novel lineages were found to be associated with mango tissues. There is a need to develop a consensus among mycologists as to which genes should be used to define and delimit a Colletotrichum species and in the mean time mycologists should voluntarily restrain from describing new species based on inadequate datasets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mango (Mangifera indica L., Anacardiaceae) is native to India where it is an important fruit crop and is now grown in most tropical and subtropical regions (Sarkiyayi et al. 2013). India is the largest producer, consumer and exporter of mango, with approximately 40 % of the total world production (Ferrier et al. 2012). Production of mango in the fiscal year 2010–11 was 15.2 million tons (Kumar et al. 2011) with India exporting 0.06 million tons of fresh mangoes to more than 30 countries worldwide and earning USD 38.2 million as revenue in the fiscal year 2011–2012 (DCGIS Annual Report 2012).
The temperature range and high humid conditions of tropical regions are favourable for anthracnose disease development, previously reported to be caused by Colletotrichum gloeosporioides sensu lato (Waller 1992; Freeman et al. 1998; Kumar et al. 2007; Laxmi et al. 2011). Colletotrichum gloeosporioides sensu lato has been reported from various tropical fruits as endophytes or pathogens and also as agents of post-harvest fruit-rots (Bailey and Jeger 1992; Freeman et al. 1998; Cai et al. 2009; Hyde et al. 2009a, b). Anthracnose disease mainly affects the aerial plant parts such as leaves, twigs, fruits and inflorescences, and causes post-harvest fruit-rots, yield and revenue loss (Swamy 2012). In addition to economic losses, issues pertaining to biosecurity of C. gloeosporioides sensu lato are also important.
Mango anthracnose is characterized by the formation of dark brown spots on fruits and leaves (Chadha 2009). Farr and Rossman (2013) listed 1481 fungal taxa associated with mango worldwide, including C. acutatum J.H. Simmonds, C. asianum Prihast. L. et al., C. capsici (Syd. & P. Syd.) E.J. Butler & Bisby (= C. truncatum (Schwein.) Andrus & W.D. Moore), C. fioriniae (Marcelino & Gouli) R.G. Shivas & Y.P. Tan, C. gloeosporioides (Penz.) Penz. & Sacc., C. horii B.S. Weir & P.R. Johnst., C. mangiferae Kelkar, C. papaya Henn. and C. simmondsii R.G. Shivas & Y.P. Tan. However, C. mangiferae and C. papaya are no longer “accepted” species of Colletotrichum (Cannon et al. 2012). Until recently (post 2008), most old records of mango anthracnose disease were based on morphological identification or similarity with the non-type sequences submitted in GenBank (Cai et al. 2011; Ko Ko et al. 2011). Thus, there is a need to reinvestigate the taxonomy of Colletotrichum taxa associated with mango anthracnose and to update existing records (Farr and Rossman 2013).
The name Colletotrichum gloeosporioides was described in Penzig (1882), based on Vermicularia gloeosporioides Penz. and it has been associated with at least 470 different host genera (Cannon et al. 2008). As host association or morphological characteristics are not reliable in distinguishing species in C. gloeosporioides sensu lato, C. gloeosporioides sensu stricto was epitypified by Cannon et al. (2008) and its usage restricted as defined by sequence-data of the 5.8S rRNA gene and flanking internal transcribed spacers 1 & 2 (ITS), the “universal” DNA barcoding marker for fungal species (Schoch et al. 2012). The ITS however, offers moderate resolution in C. gloeosporioides sensu lato (Cai et al. 2009; Crouch et al. 2009; Damm et al. 2009, 2010), thus better gene markers are needed.
Recent multigene phylogenetic analysis involving actin (act), calmodulin (cal), chitin synthase (chs1), glyceraldehyde-3-phosphate dehydrogenase (gapdh) and ITS gene regions confirmed that C. gloeosporioides sensu lato is a species complex that comprises 22 morphologically similar, phylogenetically distinct species (Weir et al. 2012). However, using a different set of gene markers (ITS, beta tubulin (tub2), DNA lyase (apn2) and the intergenic region of apn2 and MAT1-2-1 genes (ApMat)), Doyle et al. (2013) identified new lineages and described four new species within the species-complex. Presently, there are 28 accepted species names within the species complex (Peng et al. 2012; Weir et al. 2012; Doyle et al. 2013; Peng et al. 2013). There is however, no consensus among mycologists as to which gene markers should be used to define and delimit a species within the species complex (Doyle et al. 2013).
We initiated the present study to investigate the phylogenetic diversity of the C. gloeosporioides species complex associated with M. indica from India based on the 6-gene markers used by Weir et al. (2012). However, as Silva et al. (2012a) and Doyle et al. (2013) have shown that the ApMat marker provides better resolution compared to the gene-markers used by Weir et al. (2012), the ApMat marker is also incorporated in this study to resolve the species identification issues in the C. gloeosporioides species complex.
Materials and methods
Sample collection
Samples were collected from major mango growing areas in Uttar Pradesh, Delhi, Chandigarh U.T., Gujarat, Maharashtra and Goa states of India, during late April to mid July 2011. Symptomatic and asymptomatic mango fruits, leaves, twigs, shoots and inflorescences were collected from orchards, while some mango fruits with lesions were purchased from markets in Chandigarh U.T., Chennai (Tamil Nadu), Patna (Bihar), Udupi (Karnataka) and Varanasi (Uttar Pradesh) (Fig. 1).

Anthracnose symptoms on various mango host tissues: a. Young (unripe) fruits, b. Twigs and leaves, c. Green leaves and d. Mature (ripe) fruits
Fungal isolates
A total of 207 Colletotrichum isolates were included in this study. Around 600 strains were isolated from mango tissues, following the method described in Cai et al. (2009), of which 201 isolates were identified to be C. gloeosporioides sensu lato based on conidial morphology. Six Colletotrichum isolates were procured from the Microbial Type Culture Collection (MTCC), Chandigarh and Indian Type Culture Collection (ITCC), New Delhi (Supplementary Table 1). All information regarding the host substrate, variety and geographic location of these isolates are detailed in Supplementary Table 1. Available type strains of Colletotrichum species were procured from the Centraalbureau voor Schimmelcultures (CBS), Netherlands; CABI Bioscience, U.K. (formerly IMI) and Key Laboratory of Mycology at the Institute of Microbiology, Chinese Academy of Sciences, Beijing, China. The type strains of C. horii and C. xanthorrhoeae R.G. Shivas et al. were received as gift from Dr. Eric McKenzie, International Collection of Microorganisms from Plants, New Zealand. Fungal isolates were preserved at −70 °C and LN2 in 10 % glycerol for future use.
DNA extraction, PCR amplification, gene sequencing and phylogenetic analysis of the 5′tef1 gene region
Fungal isolates were subcultured on potato dextrose agar (PDA)/ potato carrot agar (PCA) (HiMedia, India) medium and grown at 20 °C for 7 days. Genomic DNA from fresh mycelia was extracted using DNA isolation kit (Zymo Research, USA; Catalogue number D6005) and stored at −20 °C. The intronic sequence of 5′ region of the translation elongation factor 1-α (5′tef1) gene region (Rojas et al. 2010) was used for selecting representative isolates for multigene phylogenetic analysis based on the terminal diversity of 207 Colletotrichum isolates. Polymerase chain reaction (PCR) was done to amplify this gene region using EF Col1- EF2 primer-set and PCR conditions as described in Rojas et al. (2010). The PCR products were cleaned using QIAquick PCR Purification Kit (QIAGEN, Catalogue number 28106) according to the manufacturer’s protocol. The purified PCR products were quantified using Nanodrop Spectrophotometer ND-1000 (Thermo) and sequenced using EF-Col1 and EF2 primers with ABI Big Dye v3.1 Terminator Ready Reaction Cycle Sequencing Kit (Applied Biosystems) using manufacturer’s protocol. The samples were purified to remove excess salt, denatured with HiDi-Formamide at 95 °C for 3 min and analyzed using 3730 DNA Analyzer (Applied Biosystems) at the central DNA sequencing facility at the Institute of Microbial Technology, Chandigarh, India.
The forward and reverse sequences obtained for each strain were aligned using Sequencher version 4.10.1 (Gene Codes Corp., Ann Arbor, Michigan, USA) to generate a consensus sequence. A multiple alignment file comprising of 226 sequences of the 5′tef1 gene region was created in MEGA version 5.05 (Tamura et al. 2011), including 207 sequences of Colletotrichum isolates from this study and 19 sequences from the type strains belonging to the C. gloeosporioides species complex retrieved from NCBI-GenBank (Supplementary Table 1 and 2). The type strain sequences generated in this study are highlighted in bold in Supplementary Table 2. Phylogenetic analysis based on Neighbour Joining (NJ) method (Saitou and Nei 1987) was performed in MEGA. The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al. 2004) and the rate variation among sites was modeled with a gamma distribution (shape parameter = 5). The resulting tree with bootstrap support for the observed branching pattern (Felsenstein 1985) is shown in Fig. 2.


a. NJ tree based on 5′tef1 gene sequences, depicting the terminal diversity of 207 Colletotrichum isolates associated with Mangifera indica from India (* = Type Strain, # = Isolate selected for multigene analysis, Clades names highlighted in red contains isolates from India, details about each clade has been mentioned in the Clade title) b. Isolates in Clade 1 c. Isolates in Clade 2 (C. jasmini-sambac) d. Isolates in Clade 7 (C. fructicola) e. Isolates in Clade 9 (C. theobromicola) f. Isolates in Clade 6 (C. asianum) g. Isolates in Clade 14 (C. siamense sensu stricto) (b–g. isolates highlighted in blue are selected for multigene phylogenetic analyses, Type Strains are highlighted in red and marked with *)
6-gene based phylogenetic analysis
Based on the 5′tef1 NJ-tree (Fig. 2), 15 Colletotrichum isolates were selected for further analysis involving act, cal, chs1, gapdh, tub2 and ITS genes. The polymerase chain reactions were carried out in an Eppendorf Mastercycler with the cycling parameters and primers as specified in previous papers (Damm et al. 2009 – ITS, act, gapdh, chs1, tub2; Weir et al. 2012 – cal). The PCR products were further processed for DNA sequencing as described above. The sequences generated in this study have been deposited in NCBI-GenBank with accession numbers as listed in Table 1.
Multigene phylogenetic analysis was carried out based on the recent 6 gene-markers used by Weir et al. (2012) to investigate the phylogenetic affinities of a subset of 15 Colletotrichum isolates within the C. gloeosporioides species complex. The four new recently described/ characterised species belonging to the C. gloeosporioides species complex (C. fructivorum V. Doyle et al., C. melanocaulon V. Doyle et al., C. rhexiae Ellis & Everh. and C. temperatum V. Doyle et al.) (Doyle et al. 2013) were not included in this analysis due to an incomplete dataset (only ITS and tub2 sequences available for these isolates). The multigene dataset was generated using SequenceMatrix version 1.7.8 (Vaidya et al. 2011). A maximum parsimony (MP) analysis of the multigene dataset was performed using PAUP version 4.0b10 (Swofford 2003). Ambiguously aligned regions were excluded from the analysis. The gaps in the alignment were treated as missing data. All the characters in the analysis were unordered and had equal weight. Trees were inferred using the heuristic search option with 20 random sequence additions and tree bisection and reconstruction (TBR) as the branch swapping algorithm. Maxtrees were set to 10,000; branches of zero length were collapsed and all multiple parsimonious trees were saved. Descriptive tree statistics [Tree Length (TL), Consistency Index (CI), Retention Index (RI), Related Consistency Index (RCI), Homoplasy Index (HI)] were calculated for the generated trees. The robustness of the trees was measured by 100 bootstrap replicates and addition of 10 random sequences. Kishino-Hasewaga tests (Kishino and Hasewaga 1989) were performed in order to determine whether trees were significantly different. Trees were viewed in Treeview (Page 1996) and edited in MEGA and Microsoft PowerPoint version 2007 (Microsoft Corp., USA). The alignment files and trees are deposited in TreeBase (www.treebase.org; Study ID: 14177). The MP tree generated using the 6-gene dataset is shown in Fig. 3.

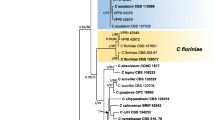
One of the 24 most parsimonious trees showing phylogenetic affinities of 15 Colletotrichum isolates associated with Mangifera indica (highlighted in blue) from India, obtained from heuristic search of the 6-gene dataset (ITS, act, cal, chs1, gapdh, tub2). C. xanthorrhoeae ICMP 17903 is the designated outgroup, and bootstrap support values more than 50 % are shown at the nodes. (Type Strains are marked with *)
ApMat-based phylogenetic analysis
Thirty-nine Colletotrichum isolates were selected for ApMat sequencing and analysis with special emphasis on their geographical origin, host-tissue type and host-variety. ApMat sequences for 161 C. gloeosporioides sensu lato isolates were retrieved from NCBI-GenBank (Please refer to Fig. 4 for more details on the host range and geographic location of these sequences). ApMat sequences for 22 type strains of the C. gloeosporioides species complex were included in the analysis (Table 2). A multiple sequence alignment for 221 taxa was used as an input file for maximum likelihood inference by GAMMA + P-Invar model of rate heteorgeneity using RaxML v. 7.2.8 server to generate 100 rapid bootstrap replicates followed by a search for the best-scoring ML tree (Stamatakis et al. 2008). The alignment files and trees are deposited in TreeBase (www.treebase.org; Study ID: 14177). The resulting ML tree is shown in Fig. 4.







Maximum likelihood tree based on ApMat dataset inferred using RaxML showing phylogenetic affinities of 39 Colletotrichum isolates associated with Mangifera indica from India with other members of C. gloeosporioides species complex C. xanthorrhoeae is the designated outgroup, and bootstrap support values more than 50 % are shown at the nodes. (Type strains are marked with *)
Morphological characterization
For selected isolates (C. asianum GM021, C. fructicola GM567, C. siamense sensu lato NK24 and C. fragariae sensu stricto GM592), morphological characterization was carried out based on the 7-day old cultures grown at 20 °C on PDA. Micro-morphological characters such as shape, size and colour of conidia and conidiogenous cells were observed and photographed using a trinocular upright microscope (Olympus U-CMAD3, Japan) equipped with an Olympus camera and differential interference contrast (DIC). For each isolate, length and width of 20 randomly chosen conidia were measured using the CellB image analysis software (Olympus, Japan). The colony diameter was measured after 7 days to determine the growth rate (mm/ day).
Pathogenicity testing
Pathogenicity testing was performed as outlined in Cai et al. (2009). Following representative isolates were selected for pathogenicity tests: C. asianum GM021, C. fructicola GM567, C. siamense sensu lato NK24 and C. fragariae sensu stricto GM592. Fresh, unripe mango fruits were surface sterilized using 1 % sodium hypochlorite solution; pin-pricked and wounded using a sterile needle. Two mangoes were inoculated with 6 μl of conidial suspension (1 × 106 spore/ml) for each isolate and one was inoculated with sterile water and used as control. The mangoes were kept in a sterile dessicator with sterile water. The appearance of disease symptoms were observed from 5–7 days of incubation at room temperature (Fig. 5). Cross infection potential of each isolates was also tested, by inoculating the conidial suspension of respective isolate to fresh green chilli fruits as described above.

Morphological features of selected Colletotrichum species and results of pathogenicity testing. a–d. Colletotrichum asianum GM021 a. Colony morphology on PDA after 5 days (front), b. Colony morphology on PDA after 5 days (reverse), c. Conidia on PDA after 7 days d. Infection symptoms on mango fruit 7 days after inoculation. e–h. Colletotrichum fructicola GM567 e. Colony morphology on PDA after 5 days (front), f. Colony morphology on PDA after 5 days (reverse), g. Conidia on PDA after 7 days h. Infection symptoms on mango fruit 7 days after inoculation. i–l. Colletotrichum siamense sensu lato NK24 i. Colony morphology on PDA after 7 days (front), j. Colony morphology on PDA after 7 days (reverse), k. Conidia on PDA after 7 days l. Infection symptoms on mango fruit 7 days after inoculation. m–p. Colletotrichum fragariae sensu stricto GM592 m. Colony morphology on PDA after 5 days (front), n. Colony morphology on PDA after 5 days (reverse), o. Conidia on PDA after 7 days p. Infection symptoms on mango fruit 7 days after inoculation (Scale bar of c, g, o = 10 μm) (Scale bar of k = 20 μm)
Results
The 5′tef1 gene-based phylogenetic diversity
The phylogenetic analysis based on 5′tef1 gene region involved 226 sequences and a total of 694 positions. All ambiguous regions were excluded from the analysis. The phylogenetic tree generated based on Neighbour Joining (NJ) method with branch support in a bootstrap analysis of 100 replicates is shown in Fig. 2. 207 Colletotrichum isolates associated with symptomatic and asymptomatic mango tissues from India clustered as six distinct clades. The Clade 1 did not contain any of the existing type strain sequence included in this study. GM390 and NK28 were selected from this clade for further multigene phylogenetic analysis. The remaining five clades consisted of the type sequences of C. asianum, C. fructicola Prihast., L. et al., C. siamense Prihast., L. et al., C. jasmini-sambac Wikee et al. and C. theobromicola. Delacr. These clades were moderately supported by the bootstrap support as shown in Fig. 2. Furthermore, GM021, GM414, GM595 were selected from the Clade 6; GM567 was selected from the Clade 7; GM592 was selected from the Clade 9; GM057, GM172, GM385, GM473, GM529, MTCC 9660, NK24 were selected from the Clade 14 and GM018 was selected from the Clade 2 for multigene phylogenetic analysis. As is evident from Fig. 2, C. jasmini-sambac, C. hymenocallidis Yan L. et al. and C. siamense sensu stricto formed separate clades in the 5′tef1 analysis.
6-gene based phylogenetic analysis
There were a total of 2675 positions in the multigene dataset. The gene boundaries in the multigene dataset included: ITS: 1–599, act: 600–891, cal: 892–1639, chs1: 1640–1938, gapdh: 1939–2245 and tub2: 2246–2675. The analysis involved 50 nucleotide sequences. Fifty characters from the ambiguous regions were excluded from the analysis. Out of the remaining 2625 characters, 2044 characters were constant, 303 characters were parsimony-informative and 278 characters were parsimony-uninformative. The maximum parsimony analysis resulted in 24 trees and based on the KH test, these trees were not significantly different (details not shown). One of the 24 trees (TL = 884, CI = 0.756, RI = 0.887, RC = 0.670, HI = 0.244) generated during the MP analysis is shown in Fig. 3. The tree was rooted with C. xanthorrhoeae ICMP 17903. The bootstrap support for the observed branching pattern is shown next to the branches.
In the MP tree shown in Fig. 3, Colletotrichum isolates GM592 clustered with the type strains of C. theobromicola (ICMP 18649, ICMP 17927, ICMP 17957) with strong bootstrap support. Colletotrichum isolates GM021, GM414, GM595 clustered with the type strain of C. asianum (ICMP 18580); GM018, GM057, GM172, GM385, GM390, GM473, GM529, MTCC 9660, NK24, NK28 clustered with the type strains of C. siamense sensu lato (ICMP 18578, ICMP 19118, ICMP 18642) while isolates GM567 grouped with type strains of C. fructicola (ICMP 18581, ICMP 18646, ICMP 17921) (Fig. 3). The observed branching pattern is similar to the species-level clades retrieved in Weir et al. (2012).
ApMat-based phylogenetic analysis
There were a total of 962 positions in the ApMat dataset. The analysis involved 221 nucleotide sequences. Fifty-eight characters from the ambiguous regions were excluded from the analysis. The final dataset comprised of 904 characters. The ML tree (rooted with C. xanthorrhoeae) having the best score (−6625.358388) is shown in Fig. 4. The ML analysis recovered the following terminal diversity: C. aotearoa B. Weir & P.R. Johnst., C. cordylinicola Phouliv. et al., C. psidii Curzi, C. clidemiae B.S. Weir & P.R. Johnst., C. rhexiae, C. fructivorum, C. kahawae J.M. Waller & Bridge / C. temperatum, C. alatae B.S. Weir & P.R. Johnst., C. gloeosporioides sensu stricto, C. salsolae B.S. Weir & P.R. Johnst., C. hymenocallidis, C. siamense sensu stricto, C. melanocaulon, C. jasmini-sambac, C. queenslandicum B.S. Weir & P.R. Johnst., C. asianum, C. tropicale E.I. Rojas et al./ C. alienum B.S. Weir & P.R. Johnst., C. musae (Berk. & M.A. Curtis) Arx, C. nupharicola D.A. Johnson et al., C. fructicola, C. horii, C. theobromicola, C. fragariae A.N. Brooks and the clades A-S which are not associated with any of the available type strains. The following species were not represented in the analysis due to non-availability of their ApMat sequences in public databases: C. aenigma B.S. Weir & P.R. Johnst., C. aeschynomenes B.S. Weir & P.R. Johnst. (Weir et al. 2012), C. murraye L.J. Peng & K.D. Hyde (Peng et al. 2012), C. ti B.S. Weir & P.R. Johnst. (Weir et al. 2012) and C. viniferum L.J. Peng et al. (Peng et al. 2013). The ApMat-based phylogeny was found to be superior in recovering terminal diversity and the observed branching received an overall good boot-strap support, except for a few branches.
Morphological comparison
The morphological characters (colony morphology on PDA, conidial measurements and shape) and growth rate comparison was made between the selected Colletotrichum isolates (C. asianum GM021, C. fructicola GM567, C. siamense sensu lato NK24 and C. fragariae sensu stricto GM592). The conidial characteristics were within the range for all four species as already described (Prihastuti et al. 2009; Rojas et al. 2010; Weir et al. 2012) (Fig. 5; Table 3).
Pathogenicity testing
The mango fruits inoculated with conidial suspension of selected Colletotrichum isolates (C. asianum GM021, C. fructicola GM567, C. siamense sensu lato NK24 and C. fragariae sensu stricto GM592) developed typical dark brown lesions of anthracnose disease around the wound. The severity of infection varied between isolates. C. siamense sensu lato NK24 produced larger lesions followed by C. asianum GM021. On the other hand, C. fructicola GM567 and C. fragariae sensu stricto GM592 produced only minor infection symptoms. The pathogen was consistently re-isolated from the infected mango fruits on to the PDA media. However, the control did not develop any symptoms after 7 days of inoculation (Fig. 5). The cross inoculation of these isolates also lead to production of disease symptoms on chilli fruits (data not shown), hinting at the broad host range of C. asianum, C. fructicola, C. siamense sensu lato and C. fragariae sensu stricto.
Discussion
Fungal systematics is facing a challenge of non-uniform application of multiple species recognition criteria (Hibbett and Taylor 2013) that has resulted in frequent, undesirable names changes, especially in economically important fungal taxa. The C. gloeosporioides species complex best represents the challenge being faced by modern mycologists as how to deal with old (now redundant) species names, while moving forward with description of new species based on multi-gene phylogenetic analysis. The name C. gloeosporioides has a long history and its nomenclatural journey since its introduction in 1882 was detailed in Cannon et al. (2008) and Weir et al. (2012), hence will not be repeated here.
In a major taxonomic development, Weir et al. (2012) reorganized the taxonomy of C. gloeosporioides species complex based on multi-gene and morphological characters. They incorporated multiple gene regions in their phylogenetic analyses but largely relied on act, cal, chs1, gapdh and ITS gene-regions to redefine species-boundaries within this species complex. Weir et al. (2012) specifically cautioned about the low levels of genetic divergence across the species complex and stressed for the need to use “powerful genes” such as ApMat and Apn25L (Silva et al. 2012a) to achieve finer resolution within the 23 species-level clades that they could recognize using limited set of genes (act, cal, chs1, gapdh, ITS).
In a recent molecular phylogenetic analysis involving the ITS, apn2, tub2 and ApMat gene-markers, Doyle et al. (2013) reported finer phylogenetic resolution in many of the previously described species of the C. gloeosporioides species complex. For example, C. siamense sensu lato (Weir et al. 2012), one of the challenging and controversial taxa, was found to be a species complex including C. jasmini-sambac, C. hymenocallidis and C. siamense sensu stricto as distinct phylogenetic species (Prihastuti et al. 2009; Yang et al. 2009; Wikee et al. 2011). Based on ApMat gene sequence-analysis, this study has identified seven distinct lineages within C. siamense sensu lato, including C. hymenocallidis, C. jasmini-sambac, C. siamense sensu stricto recently described C. melanocaulon (Doyle et al. 2013) and three un-designated clades which could potentially represent new species of Colletotrichum. Interestingly, two recently synonymised species: C. fragariae and C. theobromicola (Weir et al. 2012) also showed genetic diversity as was evident from the clustering pattern of C. fragariae and C. theobromicola into two well-supported clades (Fig. 4). We support the use of ApMat marker as an effective species identification marker for C. gloeosporioides species complex, as initiated by Silva et al. (2012a).
Mango anthracnose in India was previously reported to be caused by “C. gloeosporioides”, based on either morphology or ITS gene-sequence data (Kumar et al. 2007; Sangeetha and Rawal 2009; Gupta et al. 2010; Laxmi et al. 2011) and recently using multi-locus sequence-data (ITS and tub2) (Singh et al. 2013). It is noteworthy that none of Colletotrichum isolates associated with mango samples from India (included in this study) clustered with C. gloeosporioides sensu stricto. Thus, our study supports the previous finding by Phoulivong et al. (2010) that C. gloeosporioides sensu stricto is not a common pathogen on tropical fruits. Four known species (C. frgariae sensu stricto, C. fructicola, C. jasmine-sambac and C. melanocaulon) and five Colletotrichum lineages with no species names were found to be associated with mango tissues/ anthracnose. Our pathogenicity test results show that the representative isolates are capable of producing anthracnose symptoms on mango fruits (Fig. 5). The cross pathogenicity tests conducted using chilli fruits suggest that these isolates are not host-specific to Mangifera indica.
We have also gained an insight about host and geographical distribution of various species within the C. gloeosporioides species complex by extensive analysis involving 221 ApMat sequences (Fig. 4) as well as from other recent studies (Rojas et al. 2010; Silva et al. 2012a, b; Doyle et al. 2013). A few Colletotrichum species are restricted to a particular host, while others are limited to a particular geographical location. For example, among the major clades obtained in ApMat analysis: C. hymenocallidis is limited to China, C. aotearoa is limited to New Zealand; C. theobromicola sensu stricto is limited to Panama region; C. fructivorum, C. nupharicola and C. rhexiae are limited to North America. Details about other minor clades recovered in the ApMat analysis are provided in Table 4. Generally, members of the C. gloeosporioides complex are known to possess a broad host range, with only a few exceptions such as C. horii (Diospryos sp.) and C. musae (Musa sp.) (Table 4).
Recognition of cryptic species in a species complex with the incorporation of gene sequence-data is not limited to the C. gloeosporioides species complex and other species complexes of Colletotrichum. Many phytopathogenic genera such as Botryosphaeria (Pavlic et al. 2009; Liu et al. 2012), Diaporthe (Udayanga et al. 2011, 2012), Fusarium (Gräfenhan et al. 2011; Summerell and Leslie 2011; Summerell et al. 2011), and Mycosphaerella (Schoch et al. 2009; Hunter et al. 2011) include cryptic species that could not be diagnosed based solely on classical morpho-taxonomic characters. This warrants updating of existing checklists and fungal databases with correct taxonomic information about phytopathogenic fungal taxa to facilitate accurate plant quarantine decisions (Cai et al. 2011). In conclusion, this study has revealed the rich fungal diversity associated with different mango varieties of India. Future studies are needed to reveal cryptic species of Colletotrichum associated with other tropical fruits and also native plants. Nevertheless, consensus among mycologists as to which genes or taxonomic characters should be used while describing a fungal species will play a significant role in satisfying the needs of taxonomic end-users.
References
Bailey JA, Jeger MJ (eds) (1992) Colletotrichum: Biology, pathology and control. CAB Int, Wallingford
Cai L, Hyde KD, Taylor PWJ, Weir BS, Waller J, Abang MM, Zhang JZ, Yang YL, Phoulivong S, Liu ZY, Prihastuti H, Shivas RG, McKenzie EHC, Johnston PR (2009) A polyphasic approach for studying Colletotrichum. Fungal Divers 39:183–204
Cai L, Giraud T, Zhang N, Begerow D, Cai G, Shivas RG (2011) The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers 50:121–133
Cannon PF, Buddie AG, Bridge PD (2008) The typification of Colletotrichum gloeosporioides. Mycotaxon 104:189–204
Cannon PF, Damm U, Johnston PR, Weir BS (2012) Colletotrichum – current status and future directions. Stud Mycol 73:181–213
Chadha KL (2009) Handbook of Horticulture. Directorate of Information and Publication of Agriculture. ICAR, Krishi Anusandhan Bhavan (PUSA), New Delhi. pp 239–245
Crouch JA, Clarke BB, Hillman BI (2009) What is the value of ITS sequence data in Colletotrichum systematics and species diagnosis? A case study using the falcate-spored graminicolous Colletotrichum group. Mycologia 101:648–656
Damm U, Wouldenberg JHC, Cannon PF, Crous PW (2009) Colletotrichum species with curved conidia from herbaceous hosts. Fungal Divers 39:45–87
Damm U, Baroncelli R, Cai L, Kubo Y, O’Connell R, Weir B, Yoshino K, Cannon PF (2010) Colletotrichum: species, ecology and interactions. IMA Fungus 1:161–165
DCGIS Annual Report (2012) Agriculture and Processed food products Export Development Authority (APEDA), Ministry of Commerce and Industry, Government of India, information accessed online on 23rd April, 2013 from http://agriexchange.apeda.gov.in/product_profile/exp_f_india.aspx?categorycode=0204
Doyle VP, Oudemans PV, Rehner SA, Litt A (2013) Habitat and host indicate lineage identity in Colletotrichum gloeosporioides s. l. from wild and agricultural landscapes in North America. PLoS One 8:e62394
Farr DF, Rossman AY (2013) Fungal Databases, Systematic Mycology and Microbiology Laboratory, ARS, USDA. Retrieved April 4, 2013, from http://nt.ars-grin.gov/fungaldatabases/
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol 39:783–791
Ferrier P, Petersen E, Landes M (2012) Specialty crop access to US markets: A case study of Indian mangoes. US Department of Agriculture, Economic Research Service
Freeman S, Katan T, Shabi E (1998) Characterization of Colletotrichum species responsible for anthracnose diseases of various fruits. Plant Dis 82:596–605
Gräfenhan T, Schroers HJ, Nirenberg HI, Seifert KA (2011) An overview of the taxonomy, phylogeny and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella and Volutella. Stud Mycol 68:79–113
Gupta VK, Pandey A, Kumar P, Pandey BK, Gaur RK, Bajpai V, Sharma N, Sharma S (2010) Genetic characterization of mango anthracnose pathogen Colletotrichum gloeosporioides Penz. by random amplified polymorphic DNA analysis. Afr J Biotechnol 9:4009–4013
Hibbett DS, Taylor JW (2013) Fungal systematics: is a new age of enlightenment at hand? Nat Rev Microbiol 11:129–133
Hunter GC, Crous PW, Carnegie AJ, Burgess TI, Wingfield MJ (2011) Mycosphaerella and Teratosphaeria diseases of Eucalyptus; easily confused and with serious consequences. Fungal Divers 50:145–166
Hyde KD, Cai L, McKenzie EHC, Yang YL, Zhang JZ, Prihastuti H (2009a) Colletotrichum: a catalogue of confusion. Fungal Divers 39:1–17
Hyde KD, Cai L, Cannon PF, Crouch JA, Crous PW, Damm U, Goodwin PH, Chen H, Johnston PR, Jones EBG, Liu ZY, McKenzie EHC, Moriwaki J, Noireung P, Pennycook SR, Pfenning LH, Prihastuti H, Sato T, Shivas RG, Tan YP, Taylor PWJ, Weir BS, Yang YL, Zhang JZ (2009b) Colletotrichum – names in current use. Fungal Divers 39:147–182
Kishino H, Hasewaga M (1989) Evaluation of maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order of Hominoidea. J Mol Evol 29:170–179
Ko Ko TW, McKenzie EHC, Bahkali AH et al (2011) The need for re-inventory of Thai phytopathogens. Chiang Mai J Sci 38:625–638
Kumar AS, Reddy NPE, Reddy KH, Devi MC (2007) Evaluation of fungicidal resistance among Colletotrichum gloeosporioides isolates causing mango anthracnose in Agri Export Zone of Andhra Pradesh, India. Plant Pathol Bull 16:157–160
Kumar B, Mistry NC, Singh B, Gandhi CP (2011) Indian Horticulture Databse-2011. National Horticulture Board, Ministry of Agriculture, Government of India, New Delhi
Laxmi BKM, Reddy PN, Prasad RD (2011) Cross infection potential of Colletotrichum gloeosporioides Penz. isolates causing anthracnose in subtropical fruit crops. Trop Agric Res 22:183–193
Liu JK, Phookamsak R, Doilom M, Wikee S, Li YM, Ariyawansha H, Boonmee S et al (2012) Towards a natural classification of Botryosphaeriales. Fungal Divers 57:149–210
Page RDM (1996) TREEVIEW: an application of display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Pavlic D, Slippers B, Coutinho TA, Wingfield MJ (2009) Multiple gene genealogies and phenotypic data reveal cryptic species of the Botryosphaeriaceae: a case study on the Neofusicoccum parvum/N. ribis complex. Mol Phylogenet Evol 51:259–268
Peng LJ, Yang Y, Hyde KD, Bahkali AH, Liu Z (2012) Colletotrichum species on Citrus leaves in Guizhou and Yunnan provinces, China. Cryptogam Mycol 33:267–283
Peng LJ, Sun T, Yang YL, Cai L, Hyde KD, Bahkali AH, Liu ZY (2013) Colletotrichum species on grape in Guizhou and Yunnan provinces, China. Mycoscience 54:29–41
Penzig AGO (1882) Fungi agrumicoli. Contribuzione allo studio dei funghi parassiti degli agrumi. Michelia 2:385–508
Phoulivong S, Cai L, Chen H, McKenzie EHC, Abdelsalam K, Chukeatirote E, Hyde KD (2010) Colletotrichum gloeosporioides is not a common pathogen on tropical fruits. Fungal Divers 44:33–43
Prihastuti H, Cai L, Chan H, McKenzie EHC, Hyde KD (2009) Characterization of Colletotrichum species associated with coffee berries in northern Thailand. Fungal Divers 39:89–109
Rojas EI, Rehner SA, Samuels GJ (2010) Colletotrichum gloeosporioides s. l. associated with Theobroma cacao and other plants in Panama: multilocus phylogenies distinguish host-associated pathogens from asymptomatic endophytes. Mycologia 102:1318–1338
Saitou N, Nei M (1987) The neighbor joining method: a new method for re-constructing phylogenetic trees. Mol Biol Evol 4:406–425
Sangeetha CG, Rawal RD (2009) Temperature requirements of different isolates of Colletotrichum gloeosporioides isolated from mango. American-Eurasian J Sci Res 4:20–25
Sarkiyayi S, Mohammed M, Yakubu A (2013) Comparative analysis of nutritional and anti nutritional contents of some varieties of mango (Mangifera indica) in Kaduna Metropolis-Nigeria. Res J Appl Sci Eng Technol 5:387–391
Schoch CL, Crous PW, Groenewald JZ, Boehm EWA, Burgess TI, De Gruyter J, De Hoog GS et al (2009) A class-wide phylogenetic assessment of Dothideomycetes. Stud Mycol 64:1–15
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, André Levesque C, Chen W et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci (USA) 109:6241–6246
Silva DN, Talhinas P, Várzea V, Cai L, Paulo OS, Batista D (2012a) Application of the Apn2/MAT locus to improve the systematics of the Colletotrichum gloeosporioides complex: an example from coffee (Coffea spp.) hosts. Mycologia 104:396–409
Silva DN, Talhinhas P, Cai L, Manuel L, Gichuru EK, Loureiro A, Várzea V, Paulo OS, Batista D (2012b) Host-jump drives rapid and recent ecological speciation of the emergent fungal pathogen Colletotrichum kahawae. Mol Ecol 21:2655–2670
Singh V, Mishra RK, Mathew AJ, Pandey BK (2013) Molecular characterization of mango anthracnose pathogen Colletotrichum gloeosporioides sensu lato. Gene. doi:10.1016/j.gene.2013.05.010
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57:758–771
Summerell BA, Leslie JF (2011) Fifty years of Fusarium : how could nine species have ever been enough? Fungal Divers 50:135–144
Summerell BA, Leslie JF, Liew ECY, Laurence MH, Bullock S, Petrovic T, Bentley AR, Howard CG, Peterson SA, Walsh JL (2011) Fusarium species associated with plants in Australia. Fungal Divers 46:1–27
Swamy JS (2012) Anthracnose - a devastating pre and post-harvest disease in mango. Int J Plant Prot 5:429–437
Swofford DL (2003) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci (USA) 101:11030–11035
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Udayanga D, Liu XX, McKenzie EHC, Chukeatirote E, Bahkali AH, Hyde KD (2011) The genus Phomopsis: biology, applications, species concepts and names of common phytopathogens. Fungal Divers 50:189–225
Udayanga D, Liu XX, Crous PW, McKenzie EHC, Chukeatirote E, Hyde KD (2012) A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers 56:157–171
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180
Waller JM (1992) Colletotrichum diseases of perennial and other cash crops. In: Bailey JA, Jeger MJ (eds) Colletotrichum: Biology, pathology and control. CAB International, Wallingford, pp 131–142
Weir BS, Johnston PR, Damm U (2012) The Colletotrichum gloeosporioides species complex. Stud Mycol 73:115–180
Wikee S, Cai L, Pairin N, McKenzie EHC, Su YY, Chukeatirote E, Thi HN, Bahkali AH, Moslem MA, Abdelsalam K, Hyde KD (2011) Colletotrichum species from Jasmine (Jasminum sambac). Fungal Divers 46:171–182
Yang YL, Liu ZY, Cai L, Hyde KD, Yu ZN, McKenzie EHC (2009) Colletotrichum anthracnose of Amaryllidaceae. Fungal Divers 39:123–146
Acknowledgments
We thank Institute of Microbial Technology (CSIR-IMTECH) for the financial support. We thank Dr. Eric McKenzie from International Collection of Microorganisms from Plants, New Zealand and Dr. Makoto Kawase from National Institute Agrobiological Sciences Japan for providing some Colletotrichum type strains as gifts. Dr. D. Ananthapadmanaban, Deepak Bhatt, Amandeep Kaur, Deepinder Kaur and Yamini Agrawal from CSIR-IMTECH are thanked for their encouragement. Drs. Lei Cai and Roger Shivas are thanked for the inspiration and useful discussions on Colletotrichum taxonomy. We also thank Dr. Rakesh Pandey and Mr. Ashwin Nannaware (CSIR-NBRI, Lucknow, Uttar Pradesh), Mr. Pradip Kumar Singh (Banaras, Uttar Pradesh), Mr. Chittarjeet Singh (CSIR-CSIO, Chandigarh), Mr. Jignesh Patel (Gujrat), Mr. Srinivas Kamath (Maharashtra), Dr. S. Siddharthan (Chennai, Tamil Nadu), Ms. Sonika Verma (Patna, Bihar); Mr. Arun Prabhu and Pai (Goa) for their help in sample collections from the respective states. We acknowledge the help of Dr. V. Shanmugham, CSIR-IHBT, Palampur, India in performing the pathogenicity testing. Gavin Blyth, Landcare Research, New Zealand provided some sequences. This work was supported by CSIR-IMTECH-OLP0071 project and CSIR-SRF fellowship awarded to GS and UGC-SRF fellowship to NK. K.D. Hyde thanks the National Research Council of Thailand for the award of grant No. 54201020003 and a grant from the National Plan of Science and Technology, King Abdulaziz City of Science and Technology, Riyadh, Saudi Arabia, project No. 10-Bio-965-02 to study Colletotrichum.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
List of Colletotrichum isolates associated with Mangifera indica included in this study with information on taxon, substrate, variety, geographic location, collector’s name and GenBank accession number for the 5 ′tef 1 gene sequenced (DOC 436 kb)
Supplementary Table 2
List of the Colletotrichum type strains included in this study with information on taxon, host, geographic location and GenBank accession number for the 5′tef1 gene sequence (GenBank accession numbers highlighted in bold have been generated in this study, * = Type strain) (DOC 54 kb)
Rights and permissions
About this article
Cite this article
Sharma, G., Kumar, N., Weir, B.S. et al. The ApMat marker can resolve Colletotrichum species: a case study with Mangifera indica . Fungal Diversity 61, 117–138 (2013). https://doi.org/10.1007/s13225-013-0247-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-013-0247-4