
Abstract
Fungi of Nectriaceae are economically important and of high species diversity. For the purpose of accurate and rapid species identification, ITS, 28S rDNA, β-tubulin gene and EF-1α gene were selected as the candidate DNA barcode markers to investigate their feasibility in identification of 28 well-circumscribed species belonging to 9 genera of the nectriaceous fungi. A total of 216 sequences of the candidate genes were analyzed. Intra- and inter-specific variations and success rate of PCR amplification and sequencing were considered as important criteria to estimate the candidate genes. The partial β-tubulin gene met the requirements for an ideal DNA barcode and functions well for correct species delimitation. No overlapping between the intra- and inter-specific pairwise distances was found. The smallest inter-specific distance of β-tubulin gene was 3.45%, while the largest intra-specific distance was 2.77%; which appeared to possess the appropriate intra- and inter-specific variations. Twenty-eight clusters were recognized in accordance with the 28 morphological species tested. In addition, it had a high PCR and sequencing success rate. As to the other candidates, EF-1α gene showed fairly good sequence variations among species, but the PCR and sequencing success rate reached only 75.3%. ITS had a high PCR and sequencing success rate (93.5%) and recognizes 92.9% of the total number of species, nevertheless, overlapping occurred between the intra- and inter-specific distances, which may lead to incorrect species identification. 28S rDNA is most conservative compared with any other candidate markers and able to recognize merely 60.7% of the total species. We propose β-tubulin gene as the possible barcode for the nectriaceous fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The nectriaceous fungi (Hypocreales, Sordariomycetes, Ascomycota) are world-wide distributed, with a wide range of habitation; economically important as plant pathogens; mycoparasites; producers of antibiotics, new bioactive compounds and mycotoxins; and rarely are a source of food (Booth 1971; Rossman 1996; Rossman et al. 1999; Kirk et al. 2008; Li et al. 2009). This group shows a very high species diversity and broad range of lifestyles. There are increasing requirements of rapid species identification of the nectriaceous fungi for the purpose of plant disease diagnoses, discovery of new bioactive compounds, exploration of potential biocontrol agents, as well as protection from harmful mycotoxins.
For species identification of Nectriaceae, detailed morphology, anatomy, teleomorph−anamorph connection, perithecial reactions to KOH and lactic acid, and selected DNA sequence data are commonly required (Rehner and Samuels 1995; Rossman et al. 1999; Zhang and Zhuang 2006; Luo and Zhuang 2010), which takes a lot of effort and is time-consuming. Sometimes, two different species may share the same sequence data of the internal transcribed spacers of ribosomal RNA (ITS) and 28S rDNA (unpublished information), while morphology distinguishes them clearly. Screening an appropriate DNA barcode for efficient species identification of the nectriaceous fungi is essential.
DNA barcoding is powerful in rapid species identification for biodiversity assessment, detection of pest species invasion, food and feedstuff safety, and human health (Hebert et al. 2003a; Armstrong and Ball 2005; Ball and Armstrong 2006; Stoeckle and Hebert 2008; Valentini et al. 2009). A DNA barcode is a short, universal DNA sequence which displays greater divergences among species than those within a species (Hebert et al. 2003a, b). It has been suggested that mitochondrial cytochrome c oxidase 1 gene (COI) delivers a good species resolution and may serve as a universal barcode for major groups of animals (Hebert et al. 2003b; 2004; 2009). The combination of plastid genes rbcL and matK was adopted recently as the main barcode for land plants (Hollingsworth et al. 2009). For the Kingdom Fungi, a universal barcode has not been formally established but potential DNA barcode markers for selected taxonomic groups has been refurbished (Seifert 2009). ITS has been successfully used for species discrimination of six genera in Zygomycetes (Schwarz et al. 2006), Trichoderma and Hypocrea in Ascomycota (Druzhinina et al. 2005), and Cortinarius section Calochroi (Frøslev et al. 2007) and Melampsora (Feau et al. 2009) in Basidiomycota. But ITS appeared to be problematic in the identification of the blue stain fungi (Roe et al. 2010). For Aspergillus species diagnosis, beta (β)-tubulin gene is suggested as the potential DNA barcode locus against COI, ITS and the intergenic spacer of the ribosomal genes (IGS) (Geiser et al. 2007). Nuclear 28S rDNA and β-tubulin genes provide a perfect capability of species separation in Fusarium (O’Donnell and Cigelnik 1997). Translation elongation factor 1α gene (EF-1α) is a reliable barcode for Fusarium (Geiser et al. 2004), Trichoderma and Hypocrea (Druzhinina et al. 2005). However, our knowledge on exploration of DNA barcoding for fungi is still limited and clearly much more work is required.
Several criteria have been subjected to determination of an ideal DNA barcode, such as a short fragment, standard, universally used, having adequate variations among species and conservative within a species, exhibiting a high species resolving power (Taberlet et al. 2007; Hollingsworth et al. 2009; www.barcoding.si.edu/PDF/Guidelines for non-CO1 selection FINAL.pdf). We propose that the appropriate intra- and inter-specific sequence variations, i.e. divergent among species but relatively stable within a species (Hebert et al. 2004), and easiness of nucleotide sequence acquisition (Hollingsworth et al. 2009) are the two important aspects in evaluating feasibility of a DNA fragment as a barcode marker. Accordingly, intra- and inter-specific variations of candidate DNA barcodes for species tested, frequency distribution of intra- and inter-specific pairwise distances, clustering at a given threshold for species identification are analyzed. Undoubtedly, success rate of PCR amplification and sequencing of each gene must be taken into account for assessing the ease of nucleotide sequence acquisition.
In this study, we selected ITS, 28S rDNA, β-tubulin gene and EF-1α gene as candidate markers, which have been commonly used in fungal species identification or phylogenetic studies to investigate the possible DNA barcode for some nectriaceous fungi.
Materials and methods
Materials
A total of 216 sequences of the four candidate markers, ITS, 28S rDNA, β-tubulin gene and EF-1α gene, from 28 clearly documented and generally accepted species belonging to 9 genera of Nectriaceae were analyzed. Bionectria wenpingii was used as outgroup. Most sequences were newly provided by our research group incorporated with some retrieved from the GenBank (Table 1).
DNA amplification and sequencing
Genomic DNA of each strain was isolated from mycelium grown on potato dextrose agar (PDA) at room temperature for about 2 weeks (Wang and Zhuang 2004). The two nuclear rDNA fragments, ITS and domains D1, D2 and D3 of 28S rDNA, were amplified and sequenced with two primer pairs, ITS5 and ITS4 (or ITS1 and ITS4) (White et al. 1990), and LROR and LR5 (Vilgalys and Hester 1990; Rehner and Samuels 1994), respectively. Partial β-tubulin gene was amplified with the primers T1, T22, T222 and T224; and sequenced with T1 and Bt2b (O’Donnell and Cigelnik 1997; Glass and Donaldson 1995). Primers 526 F, 728F and 1567R were employed to amplify partial EF-1α gene segments; and the primers 526 F, 728F, EFjR and 1567Ra were used to sequence the amplicons (Carbone and Kohn 1999; Rehner and Buckley 2005; http://www.aftol.org/pdfs/EF1primer.pdf). The region between 728F and EFjR was analyzed.
PCR was performed with the Perkin Elmer GeneAmp PCR System 2400 using a 25 μL reaction system consisting of 16 μL of double distilled water, 2.5 μL of 10× PCR buffer, 2 μL of MgCl2 (25 mM), 1.25 μL of each primer (10 μM), 0.5 μL of dNTP (10 mM each), 1.25 μL of DNA template, 0.25 μL Taq DNA polymerase (5 U/μL). For ITS, PCR conditions were an initial step of 5 min at 94°C, 30 cycles of 30 s at 94°C, 30 s at 53°C, and 30 s at 72°C, followed by 10 min at 72°C. For 28 S rDNA, PCR conditions were an initial step of 5 min at 94°C, 10 cycles of 30 s at 94°C, 30 s at 62°C (decreasing 1°C per cycle), 55 s at 72°C, plus 25 cycles of 30 s at 94°C, 30 s at 52°C, 55 s at 72°C, followed by 10 min at 72°C. For β-tubulin, PCR conditions were an initial step of 5 min at 94°C, 30 cycles of 35 s at 94°C, 55 s at 53°C, 1 min or 2 min at 72°C, followed by 10 min at 72°C. For EF-1α, PCR conditions were an initial step of 5 min at 94°C, 10 cycles of 30 s at 94°C, 55 s at 63°C or 66°C (decreasing 1°C per cycle), 90 s at 72°C, plus 36 cycles of 30 s at 94°C, 55 s at 53°C or 56°C, 90 s at 72°C, followed by 7 min at 72°C. The obtained amplicons were purified by PCR Product Purification Kit (Biocolor BioScience & Technology Company) and sequencing was carried out in both directions with an ABI 3730 XL DNA Sequencer (SinoGenoMax Co. Ltd.).
Estimation of the candidate barcode markers
Comparison of intra- and inter-specific divergences
Sequences were aligned using ClustalX 1.81 (Thompson et al. 1994) and manually edited to adjust the aligned sequences by BioEdit 7.0 (Hall 1999). The aligned sequences were input into DNAstar 7.1.0 (Lasergene, WI, USA) to calculate the similarity matrices and then illustrate the intra- and inter-specific variations of the candidate barcode loci for each of the 28 investigated species in a visualization analysis tool, TaxonGap 2.4.1 (Slabbinck et al. 2008). As suggested by Martens et al. (2008), an outgroup, Bionectria wenpingii, was designated in the analyses.
The intra- and inter-specific pairwise distances were calculated using Kimura’s two-parameter (K2P) model with MEGA 4.0.2 (Tamura et al. 2007). The frequency distribution of the intra- and inter-specific distances were analyzed to check the barcoding gap, i.e. space between intra- and inter-specific distances (Meyer and Paulay 2005), with Microsoft Office Excel (Windows XP).
Another distance-dependent method using K2P model, clustering at a given threshold, for evaluation of the potential barcode regions was generated with TaxonDNA 1.6.3-dev4 (Meier et al. 2006).
Neighbor-Joining tree reconstruction
Neighbor-joining (NJ) trees were constructed using K2P model with MEGA 4.0.2 (Tamura et al., 2007) for individual candidate barcode markers to provide a graphic representation of species divergence. Bionectria wenpingii was used as outgroup.
Assessment of the easiness of test barcode sequence acquisition
The success rates of PCR amplification and sequencing of the considered DNA barcode markers for the family were assessed. A single PCR band obtained was considered as success of amplification. High quality chromatogram counted for success of sequencing. The success rate of PCR amplification multiplied by that of sequencing made the success rate of PCR amplification and sequencing.
Results
To meet the requirements for a standard DNA barcode, the sequence lengths of all the candidate markers are short. The fragments obtained are 446−502 base pairs (bp) for ITS, 479−487 bp for 28S rDNA, 472−566 bp for β-tubulin gene (including 3 introns) and 328−472 bp for EF-1α gene (including 2 introns).
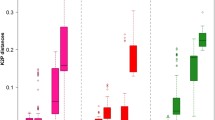
To select an appropriate or ideal DNA barcode marker, comparison of intra- and inter-specific variations is treated as a very important criterion. The comparisons of the four candidate gene markers for each of the 28 Nectriaceae species tested are performed by TaxonGap (Slabbinck et al. 2008) and the results are illustrated in Fig. 1. In general, the inter-specific variations shown by the β-tubulin and EF-1α genes were very similar and larger apparently than those of ITS and 28S rDNA. It is clear that the maximum intra-specific variations were smaller than the minimum inter-specific variations for all the species tested when the β-tubulin and EF-1α genes were applied. In contrast, the intra-specific variations of an individual species exceeded the inter-specific variations of some others in the ITS and 28S rDNA datasets, which may lead to misidentifications. In the case that ITS acted as the candidate marker, the intra-specific variations for Albnectria rigidiuscula was larger than the inter-specific variations between Gibberella acuminata and G. pulicaris. The three species may not be identified correctly. As expected, the inter-specific variations for Bionectria wenpingii, outgroup of the nectriaceous fungi, to any nectriaceous fungi tested were the largest for all the candidate genes.

Comparisons of intra- and inter-specific variations among ITS, 28S rDNA, β-tubulin and EF-1α genes of the nectriaceous fungi generated by the software TaxonGap. The grey and black bars represent the intra- and inter-specific variations respectively. The thin, black lines indicate the smallest interspecific variation. Names next to the dark bars indicate the closet species. Bionectria wenpingii was used as outgroup
The frequency distribution of intra- and inter-specific pairwise distances of the four candidate genes analyzed by MEGA and Excel is shown in Fig. 2. The inter-specific distances were much larger than the intra-specific distances for the partial sequences of the protein-coding genes (β-tubulin and EF-1α) and a distinct gap was present between intra- and inter-specific distances. Nevertheless, overlapping occurred in the datasets of ITS and 28S rDNA, which indicates that neither of them are qualified as a barcode marker.

Comparisons of frequency distribution of intra- and inter-specific pairwise distances among ITS, 28S rDNA, β-tubulin and EF-1α genes for the nectriaceous fungi generated with MEGA and Excel. The intra- and inter-specific distances are shown as white and black bars respectively
Clustering at a given threshold calculated by TaxonDNA (Meier et al. 2006) provides additional measurements of species identification success of a barcode marker. The 28 clusters produced by the β-tubulin and EF-1α genes are corresponding to the 28 well-circumscribed species of the family when the largest intra-specific distances were used as the threshold of clustering (Table 2). ITS recognized as many as 26 species, and 28S rDNA distinguished merely 17 species as a whole.
As another important criterion, success rate of PCR amplification and sequencing influences directly the efficiency of DNA barcode application. In this study, three of the four candidate genes were readily amplified and sequenced, and gained fairly high success rates (≥ 92%) except for EF-1α gene which received a relatively low rate (75.3%) (Table 3).
Almost all species were separated from each other in the four NJ trees generated from the candidate genes (Supplementary Figs. 1, 2, 3 and 4). Exception was found occasionally since Gibberella acuminata was highly cohesive with G. pulicaris in the ITS-based NJ tree. In most cases, sequences of the same species showed high cohesion, whereas, sequence divergence of the same species resulted in somewhat different tree topologies. For example, the terminal branches formed by the strains of Neonectria discophora var. discophora were not the same in the four NJ trees.
Discussion
Judged by the two important criteria for evaluating a DNA barcode marker, i.e. the suitable intra- and inter-specific variations and high success rate of PCR amplification and sequencing, our study suggests the adoption of the partial β-tubulin gene as a DNA barcode of the nectriaceous fungi.
The partial β-tubulin gene functions excellently as a barcode for the Nectriaceae species tested. Firstly, it possesses the appropriate intra- and inter-specific variations and distinguishes precisely these two types of variations. No overlapping occurred between the intra- and inter-specific pairwise distances (Fig. 2). This is substantiated by using the method TaxonGap (Slabbinck et al. 2008) where all the inter-specific variations were greater than the intra-specific ones, and where a gap existed (Fig. 1). To further confirm our conclusion, TaxonDNA was also applied (Meier et al. 2006) and the following example explains. Among Neonectria coccinea, N. ramulariae, N. shennongjiana and N. ditissima, the representatives of the genus Neonectria, the smallest inter-specific distance was 3.45% as happened between N. shennongjiana and N. ramulariae, while the largest intra-specific distance was 2.77% (Table 2).The 28 clusters match exactly the 28 species involved when the largest intra-specific distance was set up as the threshold of clustering (Table 2). This gene shows a high species identification power. Secondly, the PCR amplification and sequencing success rate of β-tubulin gene is high (92%) which is slightly lower than that of 28S rDNA (100%) (Table 3). Therefore, we recommend using the β-tubulin gene as a DNA barcode for Nectriaceae. As shown in the previous studies by other authors, it might also have the potential to become the barcode marker for other fungal groups (Glass and Donaldson 1995; Schroers et al. 2005).
EF-1α gene has shown a fairly good sequence divergence which is equally high as that observed in β-tubulin gene. All 28 species were clustered and corresponding to the 28 species tested (Table 2). This result supports the previous statements by Druzhinina et al. (2005) and Geiser et al. (2004) that EF-1α gene offers an excellent phylogenetic identification of species in the genera Trichoderma, Hypocrea and Fusarium. Unfortunately, it had a relatively poor PCR amplification and sequencing success rate (75.3%) (Table 3). It would be possible to work as a barcode marker for the family if a higher success rate of PCR and sequencing can be reached. The optimization of primer designing might be the critical point of future research.
ITS has been one of the genes routinely employed to explore the phylogenetic relationships among species of different fungal groups. ITS has also been proposed tentatively as the universal DNA barcode for the Kingdom Fungi (Seifert 2009). In this study, we found that ITS has a high PCR and sequencing success rate (93.5%) and recognizes 92.9% of the total number of species involved (Tables 2 and 3). Nevertheless, overlapping occurred between the intra- and inter-specific distances. As calculated by TaxonDNA (Meier et al. 2006), the largest intra-specific pairwise distance was up to 1.30%, whereas the inter-specific pairwise distances between strains of Gibberella acuminata and that of G. pulicaris were only 0% and 0.21%. In another case, the inter-specific pairwise distance between Neonectria ramulariae and N. shennongjiana were 1.36% which is almost the same as the highest intra-specific distance (Figs. 1 and 2) (Table 2). The overlapping and failure of accurate identification restrict the use of ITS as a barcode marker for Nectriaceae even though it provides a relatively good species discrimination.
The nuclear large subunit ribosomal RNA gene (LSU) was commonly employed for investigations of phylogenetic relationships among certain groups of fungi at generic or a higher taxonomic level (Johnson and Vilgalys, 1998). It was reported to be suitable for species recognition in yeasts (Kurtzman and Robnett 1998; Fell et al. 2000; Ninet et al. 2003) and thus being considered as a barcode of yeasts (Seifert 2009). Our study shows that 28S rDNA (LSU) is the most conservative marker compared with any other candidate markers and that its sequence divergences are not sufficient to distinguish species accurately. In the case of Neonectria species, N. coccinea, N. shennongjiana, N. ramulariae and N. ditissima are grouped together as a single cluster when performed with TaxonDNA (Meier et al. 2006). Seventeen clusters are corresponding to the 28 taxonomic species, i.e. recognizing merely 60.7% of the total number of species tested. The inadequate intra- and inter-species variations make 28S rDNA fail to be a barcode for Nectriaceae.
References
Armstrong KF, Ball SL (2005) DNA barcodes for biosecurity: invasive species identification. Phil Trans R Soc B 360:1813–1823
Ball SL, Armstrong KF (2006) DNA barcodes for insect pest identification: a test case with tussock moths (Lepidoptera: Lymantriidae). Can J For Res 36:337–350
Booth C (1971) The genus Fusarium. Commonwealth Mycological Institute, Kew, Surrey
Carbone I, Kohn LM (1999) A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91:553–556
Druzhinina IS, Kopchinskiy AG, Komoń M, Bissett J, Szakacs G, Kubicek CP (2005) An oligonucleotide barcode for species identification in Trichoderma and Hypocrea. FungGenet Biol 42:813–828
Feau N, Vialle A, Allaire M, Tanguay P, Joly DL, Frey P, Callan BE, Hamelin RC (2009) Fungal pathogen (mis-) identifications: A case study with DNA barcodes on Melampsora rusts of aspen and white poplar. Mycol Res 113:713–724
Fell JW, Boekhout T, Fonseca A, Scorzetti G, Statzell-Tallman A (2000) Biodiversity and systematics of basidiomycetous yeasts as determined by large-subunit rDNA D1/D2 domain sequence analysis. Int J Syst Evol Microbiol 50:1351–1371
Frøslev TG, Jeppesen TS, Læssøe T, Kjøller R (2007) Molecular phylogenetics and delimitation of species in Cortinarius section Calochroi (Basidiomycota, Agaricales) in Europe. Mol Phylogenet Evol 44:217–227
Geiser DM, Jiménez-Gasco M, Kang S, Makalowska I, Veeraraghavan N, Ward TJ, Zhang N, Kuldau GA, O’Donnell K (2004) FUSARIUM-ID v.1.0: A DNA sequence database for identifying Fusarium. Eur J Plant Pathol 110:473–479
Geiser DM, Klich MA, Frisvad JC, Peterson SW, Varga J, Samson RA (2007) The current status of species recognition and identification in Aspergillus. Stud Mycol 59:1–10
Glass NL, Donaldson GC (1995) Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microb 61:1323–1330
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hebert PD, Cywinska A, Ball SL, de Waard JR (2003a) Biological identifications through DNA barcodes. Proc R Soc Lond 270:313–321
Hebert PD, Ratnasingham S, de Waard JR (2003b) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B (Suppl) 270:S96–S99
Hebert PD, Stoeckle MY, Zemlak TS, Francis CM (2004) Identification of Brids through DNA Barcodes. PLoS Biol 2:e312
Hebert PD, de Waard JR, Landry J-F (2009) DNA barcodes for 1/1000 of the animal kingdom. Biol Lett. doi:10.1098/rsbl.2009.0848
Hollingsworth PM, Forrest LL, Spouge JL et al (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106:12794–12797
Johnson J, Vilgalys R (1998) Phylogenetic systematics of Lepiota sensu lacto based on nuclear large subunit rDNA evidence. Mycologia 90:971–979
Kirk PM, Cannon PF, Minter DW, Stalpers JA (eds) (2008) Dictionary of the Fungi (10th ed). CAB International. Wallingford, UK
Kurtzman CP, Robnett CJ (1998) Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26 S) ribosomal DNA partial sequences. Antonie van Leeuwenhoek 73:331–371
Li J, Liu SC, Niu SB, Zhuang WY, Che YS (2009) Pyrrolidinones from the ascomycete fungus Albonectria rigidiuscula. J Nat Prod 73:2184–2187
Luo J, Zhuang WY (2010) Three new species of Neonectria (Nectriaceae, Hypocreales) with notes on their phylogenetic positions. Mycologia 102:142–152
Martens M, Dawyndt P, Coopman R, Gillis M, De Vos P, Willems A (2008) Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int J Syst Evol Microbiol 58:200–214
Meier R, Shiyang K, Vaidya G, Ng PK (2006) DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Syst Biol 55:715–728
Meyer CP, Paulay G (2005) DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol 3:e422
Ninet B, Jan I, Bontems O, Léchenne B, Jousson O, Panizzon R, Lew D, Monod M (2003) Identification of dermatophyte species by 28 S ribosomal DNA sequencing with a commercial kit. J Clin Microbiol 41:826–830
O’Donnell K, Cigelnik E (1997) Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol Phylogenet Evol 7:103–116
Rehner SA, Samuels GJ (1994) Taxonomy and phylogeny of Gliocladium analyzed from nuclear large subunit ribosomal DNA sequences. Mycol Res 98:625–634
Rehner SA, Samuels GJ (1995) Molecular systematics of the Hypocreales: A teleomorph gene phylogeny fad the status of their anamorph. Can J Bot 73(suppl I):S816–S823
Rehner SA, Buckley E (2005) A Beauveria phylogeny inferred from nuclear ITS and EF1-α sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97:84–98
Roe AD, Rice AV, Bromilow SE, Cooke JEK, Sperling FAH (2010) Multilocus species identification and fungal DNA barcoding: insights from blue stain fungal symbionts of the mountain pine beetle. Mol Ecol Res doi:. doi:10.1111/j.1755-0998.2010.02844.x
Rossman AY (1996) Morphological and molecular perspectives on systematics of the Hpocreales. Mycologia 88:1–19
Rossman AY, Samuels GJ, Rogerson CT, Lowen R (1999) Genera of Bionectriaceae, Hypocreaceae and Nectriaceae (Hypocreales, Ascomycetes). Stud Mycol 42:1–248
Schroers H-J, Geldenhuis MM, Wingfield MJ, Schoeman MH, Yen Y-F, Shen W-C, Wingfield BD (2005) Classification of the guava wilt fungus Myxosporium psidii, the palm pathogen Gliocladium vermoesenii and the persimmon wilt fungus Acremonium diospyri in Nalanthamala. Mycologia 97:375–395
Schwarz P, Bretagne S, Gantier J-C, Garcia-Hermoso D, Lortholary O, Dromer F, Dannaoui E (2006) Molecular identification of zygomycetes from culture and experimentally infected tissues. J Clin Microbiol 44:340–349
Seifert KA (2009) Progress towards DNA barcoding of fungi. Mol Ecol Resour 9(Suppl 1):83–89
Slabbinck B, Dawyndt P, Martens M, De Vos P, De Baets B (2008) TaxonGap: a visualisation tool for intra- and inter-species variation among individual biomarkers. Bioinformatics 24:866–867
Stoeckle MY, Hebert PD (2008) Barcode of life. Sci Am 299(82–86):88
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, Vermat T, Corthier G, Brochmann C, Willerslev E (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35:e14
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Valentini A, Pompanon F, Taberlet P (2009) DNA barcoding for ecologists. Trends Ecol Evol 24:110–117
Vilgalys R, Hester M (1990) Rapid genetic identification and mapping enzymatically amplified ribosomal DNA from several Cryptococcus species. J Bacteriol 172:4238–4246
Wang L, Zhuang WY (2004) Designing primer sets for amplification of partial calmodulin genes from penicillia. Mycosystema 23:466–473
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: A Guide to Methods and Applications. Academic, New York, pp 315–322
Zhang XM, Zhuang WY (2006) Phylogeny of some genera in Nectriaceae (Ascomycetes, Hypocreales) inferred from rDNA 28 S partial sequences. Mycosystema 25:15–22
Acknowledgements
The authors express their deep thanks to Prof. R. P. Korf for corrections of the language, and Dr. R. Meier for providing a useful reference. This project was supported by the State 863 Project (no. 2008AA02Z312) and Special Project for Fundamental Research (no. 2006FY120100) from the Ministry of Science and Technology of China, and the National Natural Science Foundation of China (no. 31070015) to WYZ.
Author information
Authors and Affiliations
Corresponding author
Additional information
Peng Zhao and Jing Luo contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Suppl. Fig. 1
Neighbor-joining tree based on ITS sequences from 28 species of Nectriaceae with Bionectria wenpingii as outgroup. TreeBase S10405 (GIF 163 kb)
Suppl. Fig. 2
Neighbor-joining tree based on 28 S rDNA sequences from 28 species of Nectriaceae with Bionectria wenpingii as outgroup. TreeBase S10406 (GIF 190 kb)
Suppl. Fig. 3
Neighbor-joining tree based on β-tubulin gene sequences from 28 species of Nectriaceae with Bionectria wenpingii as outgroup. TreeBase S10407 (GIF 173 kb)
Suppl. Fig. 4
Neighbor-joining tree based on EF-1α gene sequences from 28 species of Nectriaceae with Bionectria wenpingii as outgroup. TreeBase S10408 (GIF 184 kb)
Rights and permissions
About this article
Cite this article
Zhao, P., Luo, J. & Zhuang, WY. Practice towards DNA barcoding of the nectriaceous fungi. Fungal Diversity 46, 183–191 (2011). https://doi.org/10.1007/s13225-010-0064-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-010-0064-y