
Abstract
A strain isolated from the oil reservoir in northern China was identified as Geobacillus pallidus by 16S rRNA gene sequencing. It could grow at a temperature of 45–80°C and salinity of 0–15% (w/v) and synthesize biosurfactant by using crude oil as sole carbon source under aerobic or anaerobic conditions. The yields of biosurfactant were ≈ 9.8 g/L and ≈ 2.8 g/L, respectively. Compositional analysis revealed that the fractionated components and compositions of the purified biosurfactant differed between aerobic (glycosides ≈ 50.3%, lipids ≈ 34.5%, peptide ≈ 15.2%, w/w) and anaerobic (glycosides ≈ 53.8%, lipids ≈ 31.2%, peptide ≈ 26.0%, w/w) conditions. The critical micelle concentrations of aerobic and anaerobic biosurfactant were 0.016 g/L and 0.022 g/L, respectively. Gas chromatography analysis indicated that strain H9 had a preference for utilizing medium- and long-length alkanes (C23–C43) under aerobic conditions, and degrading long alkanes (C33–C43) under anaerobic conditions. Physical simulation results showed that strain H9 and its biosurfactant have great potential use in microbial enhanced oil recovery, especially in high temperature and salinity oil reservoirs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Microbial enhanced oil recovery (MEOR) uses microorganisms and their metabolites to increase oil recovery. Oil remaining after water flooding is entrapped in pores by capillary forces. This residual oil is the main target in the application of the enhanced oil recovery (EOR) technique. MEOR is considered as an EOR procedure that is developed for use either simultaneously with water flooding or as a tertiary phase in the recovery process (Jenneman 1989). A variety of metabolites produced by the microbes naturally associated with oil fields are known to improve oil drainage into the well. Through different mechanisms, these microbial products change oil reservoir characteristics such as the wettability of the reservoir rock, or the degree of fluid saturation (McInerney and Sublette 2002; Brown 2010). The gradual alteration of the reservoir environment due to these types of biochemical events is considered a useful tool for oil recovery. The indigenous bacteria in an oil field thus play a key role in conducting a MEOR process successfully.
Surfactants—amphipathic molecules that have both hydrophilic and hydrophobic moieties—tend to partition preferentially at the interface between liquid phases with varying degrees of polarity and hydrogen-bonding characteristics such as oil–water and water–air interfaces (Desai and Banat 1997). Surfactants accumulate at interfaces between phases of different polarity and govern the arrangement of liquid molecules. This arrangement affects the formation of H-bonds and hydrophobic–hydrophilic interactions. This dynamic behavior is based on the ability of the surfactant to reduce the surface tension. Clostridium pasteurianum produces an extracellular neutral lipid that reduces the surface tension of water from 72 to 55 mN/m (Cooper and Goldenberg 1987). Youssef et al. (2007) found that Bacillus licheniformis JF-2 isolated from oil field injection water was able to grow under anaerobic conditions and produced a biosurfactant able to reduce the surface tension of the medium to less than 30 mN/m. The potential utility of this bacterium for in situ MEOR has been discussed (McInerney and Sublette 2002). In addition, a lipopeptide surfactant produced by thermophilic and halotolerant B. licheniformis BAS50 was characterized, and the purified biosurfactant was found to decrease the surface tension of water to 28 mN/m (Yakimov et al. 1995).
The physical and chemical characteristics of an oil reservoir are important parameters that place considerable restrictions on the performance of the microbes present in the oil reservoir. Temperature and salinity are among the most influential parameters. Researchers have found that there is a negative relationship between the ability of these organisms to grow at high salt concentrations and their growth ability at high temperatures (Brown 2010). A variety of bacteria exist in different environments, particularly in oil field brines, and the importance of these indigenous bacteria for the MEOR process has been discussed rather extensively (Rojo 2009). The differences in metabolic pathways between aerobic and anaerobic alkane biodegradation are well understood (Zengler et al. 1999; Chayabutra and Ju 2000; Salminen et al. 2004; Rojo 2009). This diversity could change the physical-chemical characteristics of biosurfactants and crude oil, resulting in distinct surface tension, emulsification activity, cell surface hydrophility and oil recovery efficiency. However, in addition to these characteristics, these microorganisms must be able to grow and metabolize in the hot or high salinity conditions in the reservoir. A vast array of thermophilic bacteria has been reported in the literature (Hao et al. 2004), but thermophilic and halotolerant bacteria, which could produce biosurfactants by using crude oil, have been relatively seldom reported, especially under simultaneous aerobic and anaerobic conditions.
This paper describes a biosurfactant-producing bacterium isolated from oil-containing brine from Daqing oil reservoir in China, and demonstrates its ability to biodegrade petroleum hydrocarbons under aerobic and anaerobic conditions. In addition, the characteristics (such as surface tension, emulsification and critical micelle concentration) of the purified biosurfactant produced by the strain in the two conditions were estimated. These studies aimed to provide some theoretical and experimental bases for the application of this strain and its biosurfactant in petroleum recovery. Physical simulation tests were employed to evaluate the efficiency of oil recovery.
Materials and methods
Sampling procedure
The Daqing oilfield is located in the Northwest of China. The reservoir is located at depths of 1,300–1,600 m subterranean with a temperature of about 58–68°C. The parameters of the Daqing oil reservoir are as follows: air permeability 575 × 10−3 μm2; porosity 34.7%; crude oil viscosity 79.1 mPa·s; crude oil density 0.898 g/cm3; total salinity 8,052.26 mg/L. Samples of brine and crude oil were collected from an operating LHP wells in the DaQing oil reservoir in March 2010. The samples were taken in sterile bottles and transported to the laboratory at 4°C, and bacteriological analyses were conducted as soon as possible. The chemical/physical characteristics of the brine are shown in Table 1.
Isolation of microorganisms
The brine (20 mL) and crude oil (10 g) were transferred to a 500-mL flask containing 80 mL minimal medium (MM) containing (NH4Cl 2 g/L, K2HPO4 1.5 g/L, KH2PO4, 1.5 g/L, MgCl2·6H2O 0.5 g/L, yeast extract 0.3 g/L, pH 6.8-7.2) supplemented with trace salt solution (0.1%, v/v) that contained (MnCl2·4H2O 0.1 g/L; CoCl2·6H2O 0.17 g/L; CaCl2·2H2O 0.02 g/L; FeCl2 0.4 g/L; H2BO3 0.019 g/L; ZnCl2 0.1 g/L; NaMoO4·2H2O 0.1 g/L; vitamin B12 0.0005 g/L; NiCl2·6H2O 0.05 g/L) and incubated at 65°C, 180 rpm for 20 days. Samples (0.1 mL) of the culture were taken and spread on LB nutrient agar plates and incubated for 48 h at 65°C. Pure cultures of morphologically distinct colonies were obtained by repetitive streaking on solid LB agar medium. To isolate strains capable of biosurfactant production, an overnight seed culture of each distinct isolate inoculated in 80 mL of biosurfactant-producing medium (BPM) (NaNO3 4.5 g/L, K2HPO4 1.5 g/L, KH2PO4 1.5 g/L, MgCl2·6H2O 0.5 g/L, pH 6.8–7.2) in a 250 mL Erlenmeyer flask at 65°C and 180 rpm for 20 days using paraffin (5 g/L) as carbon source. Cell-free samples from this culture broth were screened for biosurfactant production by the oil spreading method (Robert et al. 1989; Pruthi and Cameotra 2003; Afshar et al. 2008). Among several bacterial strains isolated, a potent biosurfactant-producing bacterial strain was isolated after the formation of significant clear zone on the oil surface; this strain was maintained on LB agar medium at 4°C. Chemicals used in the medium were of analytical grade and purchased from Beijing Chemical Industry Co. (Beijing, China).
Characterization of bacterial strain
The selected bacterial isolate was examined using standard biochemical tests (morphology and biochemistry) following Bergey’s manual of systematic bacteriology (Garrity et al. 2004). For 16S rRNA gene amplification and sequencing, the chromosomal DNA of strain H9 was extracted using a soil DNA Kit (Omega Biotek, Norcross, GA) following the manufacturer’s instructions. The 16S rRNA gene was then amplified by polymerase chain reaction (PCR) using one set of primers (27f,5′-AGAGTTTGATCTGGCTCAG-3′)/(1541r,5′-AAGGAGGTGATCCAGGCC-3′) (Winker and Woese 1991). The reaction was carried out in a 25 μL volume containing 1 x PCR buffer, 1.5 mmol/L MgCl2, 2 mmol/L dNTP mixture, 0.5 μmol/L each primers, 0.5 μL pfu enzyme and 1 ng template DNA. PCR amplification was performed as follows: initial denaturation at 94°C for 5 min, followed by 30 cycles each of 94°C for 1 min, 55°C of annealing for 45 s, and 72°C of extension for 90 s, and finally with 72°C of extension for 10 min. The PCR product amplified from the bacterial isolate which appeared as a single band was purified using a High Pure PCR product Purification Kit (Roche Applied Science, Roche Applied Science, Penzberg, Germany) and sequenced with an ABI Prism 377 automatic sequencer (Applied Biosystems, Foster City, CA) using primers 27f and 1541r. Sequence homologies were examined using BLAST version 2.2.12 of the National Center for Biotechnology Information (Altschul et al. 1990). Multiple sequence alignments were carried out using Clustal X, and a consensus neighbor-joining tree was constructed using Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0 (Tamura et al. 2007).
Bacterial growth at extreme conditions
Growth studies were carried out in both aerobic and anaerobic conditions in 100 mL LB medium at different temperatures (20–80°C) and NaCl concentrations (0–25%, g/L) at 200 rpm. The modified Hungate technique was used to prepare anaerobic conditions with a solution containing 2.5% (2.5g/100ml) cysteine hydrochloride and 2.5% (2.5g/100ml) Na2S·9H2O as the reducing agents (Miller and Wolin 1974; Sanchez et al. 1992). Experiments were performed in triplicate and the results are averages of three independent experiments. Culture turbidity was determined by measuring optical density at 600 nm (OD600) in 4 mL cuvettes using a spectrophotometer (Jenway Series 9100 UV).
Aerobic and anaerobic degradation
Time-course studies of two culture conditions were carried out by determination of biosurfactant activity and productivity, surface tension measurements and bacterial growth rate. LB medium (20 mL) was inoculated with a single colony of selected isolate and incubated at 60°C, 180 rpm for 18 h as the inoculum. The cells were collected by centrifugation at 12,000 g for 15 min and washed twice in 20 mL sterile phosphate buffer (K2HPO4 3.4 g/L, NaOH 7 g/L, pH 7.2). Then, 20 mL bacterial suspensions (OD600 = 1) was transferred to a 500 mL triangular flask containing 10 g sterile crude oil-containing mineral medium, and incubated at 65°C and 180 rpm for 100 days under aerobic and anaerobic conditions, respectively, with sterile cell-free medium as controls. Anaerobic conditions were prepared as follows: the medium was boiled for 30 min in order to dispel all dissolved oxygen prior to use; l-cysteine and resazurin as oxygen indicators were added to the medium to final concentrations of 0.05% and 0.01 (g/L), respectively. The anaerobic culture bottles were sealed with rubber caps, sterilized, and filled with filter-sterilized pure nitrogen gas until the oxygen indicator in the medium become achromatic. Time course samples of culture medium were drawn in appropriate intervals of time and monitored for kinetic parameters, including growth rate, surface activity indices [surface tension (ST) and emulsification activity], and biosurfactant production. Bacterial cell growth was monitored by measuring the dry cell weight. Dry cell weight was determined by centrifugation (12,000 g, 20 min) of a 10 mL culture broth, and cell pellets were washed with distilled water twice and dried by heating at 40°C until constant weight was attained. The surface tension of cell-free samples was measured by the digital tensiometer (KRUESS klot) using the ring detachment method.
The emulsification index (EI24) was evaluated according to Cooper and Goldenberg (1987) with minor modifications. For this purpose, cell-oil-free supernatant was collected by centrifugation (12,000 g, 20 min) of the culture broth. Then, 4 mL supernatant was mixed with 6 mL kerosene in a test tube. The mixture was stirred vigorously for 5 min and then left to stand for 24 h prior to measurement. The EI24 is defined as the height of the emulsion layer divided by the total height, multiplied by 100 and expressed as a percentage.
Cell surface lipophilicity (CSL) was measured by the BATH test (Rosenberg 1984). Bacterial cells were harvested separately by centrifugation (12,000 g, 20 min) and washed three times with phosphate buffer (K2HPO4 g/L, NaOH 7 g/L, pH 7.2) to remove any residual medium and extracellular polymeric substances. The cells were then re-suspended in a buffered salts solution (pH 7.0) containing 3.4 g K2HPO4, 1.5 g KH2PO4, and 0.2 g MgSO4·7H2O per liter. The 4 mL samples of a cell suspension were transferred to individual test tubes (diameter 1.7 cm; length 15 cm), each of which contained 1 mL hexadecane. The test tubes were vortexed at full speed for 5 min and then left to stand for 20 min to allow phase separation. Lipophilicity (expressed as a percentage) was calculated as follows: [1 − (a/b)] × 100 %, where a is the initial optical density (OD600) of bacterial suspension in the aqueous phase and b is the optical density (OD600) in the aqueous phase after partitioning. The mean percentage of partitioning of an organism into the hexadecane phase was calculated using triplicate samples.
Isolation of biosurfactant
The residual crude oil was centrifuged and removed from the collected supernatant during the incubation by a mixture of n-hexane chloroform and dichloromethane (1:1:1, v/v/v). Three solvent systems, i.e., a mixture of chloroform-ethanol (2:1, v/v), cold acetone and ethyl acetate, were examined for biosurfactant extraction to find the best method for efficient biosurfactant extraction. For biosurfactant extraction, the cell-oil-free supernatant was acidified to pH 2.0 using 6 mol/L hydrochloric acid solution and then kept at 4°C overnight (Cooper and Goldenberg 1987). A yellow–white biosurfactant precipitate formed and collected by centrifugation (12,000 g, 20 min) and washed several times with acidic water (pH 2.0 with 6 N HCl), dissolved in alkaline water (pH 8.0 with 1 mol/L NaOH), re-acidified and re-centrifuged at 4°C (12,000 g, 20 min). The precipitate was re-suspended in a chloroform-ethanol (2:1, v/v) combination, mixed vigorously and centrifuged (12,000 g, 20 min) to accelerate phase separation. The mixture of precipitate and chloroform-ethanol formed three phases, with the upper phase holding water and residual culture broth, and the middle phase (which was a dark-colored layer with greenish pigments) being separated and transferred to a clean bottle and extracted with chloroformethanol three times for complete recovery of the biosurfactant. The lower phase containing biosurfactant was collected in a clean evaporating dish and the solvent removed with the aid of a rotary evaporator under reduced pressure. The dried product was washed with absolute ethanol three times to complete remove of residual pigments before being lyophilized and stored for further analysis. The experiments were carried out in duplicate and the results reported are the average of two independent experiments.
Analysis of partially purified biosurfactant
The total carbohydrate content of partially purified biosurfactant was assayed using the anthrone sulfuric acid method using glucose as standard (Dubois et al. 1956; Al-Tahhan et al. 2000). A biosurfactant solution of 10 mg/mL was prepared, of which 1.0 mL was mixed with the 4 mL 0.2% (2g/100ml) anthrone-H2SO4 (98%) solution in thick-well glass test tubes with slight shaking in the ice water. Samples were left to stand for 15 min at room temperature. The glycosides concentration was measured against glucose standards by absorbance at 490 nm. Protein content was determined by the Lowry method (Lowry et al. 1951) using bovine serum albumin (BSA) as a calibration standard. The lipopenic content of extracted biosurfactant was determined by dichloromethane-methanol method described by Manocha et al. (1980) and Han and Duan (1998).
Analysis of crude oil
To assess the degradation of crude oil during 100 days incubation under aerobic and anaerobic conditions, crude oil was extracted from the supernatant by a mixture of n-hexane chloroform and dichloromethane (1:1:1, v/v/v) to calculate the degrading-ratio of crude oil at 4°C. The collected oil samples were separated into saturated hydrocarbons, aromatic hydrocarbons, non-hydrocarbons, resin and asphaltenes using column chromatography with several different developing solvents, and then weighted to evaluate the degrading-ratio as described by Yan and Xie (2009). In order to investigate further differences in crude oil degradation, saturated hydrocarbons and aromatic hydrocarbons were analyzed by gas chromatography/mass spectroscopy followed published procedures described by Reddy and Quinn (1999). The rheological studies of crude oil were carried out as described by Etoumi (2007). A rotating viscometer (CV100) set at 65°C and 12 s−1 shear rate, and differential scanning calorimetry were used to estimate the viscosity of crude oil, and wax appearance temperature (WAT) was determined as described by Li and Zhang (2003).
Physical simulation experiments in oil recovery
To evaluate the potential application of the strain H9 in EOR, the core flooding system was employed. Standard core flooding equipment similar to that described by Lotfabada et al. (2009) was used. Three stainless steel columns of 200 mm in length and 25 mm in diameter, named 1#, 2# and 3#, were packed with sandstone. The sandstone, which had a particle size distribution of 380 μm (m/m, 35%), 120–109 μm (m/m, 30%), and 80–75 μm (m/m, 35%), was free of organic contaminants, and dried for 24 h at 120°C and used throughout the study. Operation of oil recovery simulation was done as follows:
-
(1)
All columns were sterilized at 121°C for 2 h. Each sandstone core was vacuumized for about 2 h. After this operation, the cores were saturated with LHP brine for porosity and permeability determination.
-
(2)
Saturation of the sand pack with brines: the column packed with sand was vacuumized, and nitrogen gas was passed through one end for 5 min, using a syringe to remove oxygen from the column. To ensure removal of all the gases from the column, the flow of nitrogen was stopped and vacuum was held for 2 min at the other end of the column. The column was then flooded with brine at a pressure of 8–10 kg/cm2. The pore volume )PV) of the column was calculated by measuring the volume of brine required to saturate the column. Subsequently, three PVs of brine were passed through the column to ensure 100% saturation with brine.
-
(3)
Saturation of the sand pack with oil: LHP oil, heavy oil (density 898 g/L) from the Daqing oil reservoir was used in all experiments. The oil, filled in a tank, was passed under pressure into the sand pack column, in the same way as brine, until residual brine saturation was reached. As oil entered the column, brine was displaced and discharged from the pack through tubing inserted into the bottom end of the column. Initial oil saturation was calculated by measuring the volume of brine displaced by oil saturation.
-
(4)
Brine flooding (the first water flooding): the sand pack was again flooded with brine until there was no oil coming in the effluent, i.e., residual oil saturation was reached. Approximately six to nine PVs of brine were sufficient to reduce the pack nearly to its residual oil saturation. The amount of crude oil retained in the sand pack was determined volumetrically. Residual oil saturation was calculated by measuring the volume of displaced oil.
-
(5)
Strain flooding under two patterns: this was done in a manner similar to oil and brine floods. 0.5 PV of MS medium and H9 strain was passed through the #1 and #2 column at a flow rate of approximately 2.0 mL/min and incubated for 100 days with or without air (0.5 PV) to format aerobic condition. #3 was a control experiment with no strain, nutrition activator and air. Then, the columns were again flooded with LHP brine (the second water flooding). Discharges from the column were collected in 25 mL quantities to measure the amount of oil recovered. These experiments were used to check their oil recovery efficiency and done in triplicate, and the results presented were average data. The basic parameters of the model reservoirs are listed in Table 2.
Table 2 Basic parameters of the testing core model. Data are average values of three parallel experiments and the standard deviation is 0.1 ± 0.02
Results and discussion
Characterization of bacteria
The complete sequence of the strain was determined, which amounts to more than 99% of the 16S rRNA gene. The DNA base composition of strain H9 was around 52.6% mol G + C. In the phylogenetic analysis, the sequence showed 99.0% sequence similarity with Geobacillus pallidus. The phylogenetic relationship of the strain is shown in Fig. 1. The sequence of the 16S rRNA gene of strain H9 has been deposited with GenBank and is available under the accession number FJ788895.1.

Phylogenetic tree of Geobacillus pallidus H9 based on the 16S rDNA sequence constructed by the neighbor-joining method, demonstrating the phylogenetic position of strain H9 within the radiation of Geobacillus pallidus numbers represent confidence estimates determined by bootstrap analysis with 1,000 replicates
This strain is a facultative anaerobic, Gram-positive rod-shaped spore-forming bacterium, non-motile, no flagellum with a length of 1.9–3.6 μm and a width of 0.6–0.9 μm. Colonies were yellow and round with a translucent halo and flattened out with 4 days incubation. The sporangium was not swollen, whereas the spores were oval and subterminal positioned. Strain H9 used a variety of carbon sources for growth, including l-arabinose, fructose, galactose, glucose, inulin, d-xylose, d-mannoal, salicin, and lactose but not adonitol, melibiose. Catalase and H2S tests were positive. The strain reduced nitrate and did not produce indole or hydrogen sulfide. Arginine dehydrolase and β-galactosidase activities were observed, but no activities of oxidase, ornithine decarboxylase, lysine decarboxylase, phenylalanine desaminase, or tryptophan desaminase were detected.
Bacterial growth at extreme conditions
The key parameters for use of bacteria for enhancing oil recovery purposes are salinitiy and temperature. The results obtained from the two culture patterns under the different temperatures are shown in Fig. 2a. The maximum optical density of isolate H9 under aerobic conditions was 1.438 at the temperature of 65°C, and 1.12 under anaerobic conditions when the temperature of the culture was 70°C. Below 70°C, the optical density of the aerobic culture was higher than that of the anaerobic culture, but the trend was opposite after 70°C. And the adaptive range of temperature was 40–80°C (aerobic) and 50–80°C (anaerobic). The tolerance to NaCl levels decreased in the light of the three phases as shown in Fig. 2b. The growth trend of strain H9 at different salinities was similar under the two conditions. From 0–3% NaCl, the optical density (OD) increased slowly and no negative influence on cell growth was observed. However, from 3–8% NaCl, OD decreased, and the decreasing-rate of aerobic culture was higher than that under anaerobic conditions, while from 8–15% NaCl, the decreasing-rate of anaerobic culture increased and was more than that under aerobic conditions. From 15–25% NaCl, the isolate growth under the two conditions was non-significant. Based on these results, isolate H9 was identified as a halotolerant and thermophilic bacterium. However, further research into the reasons for the differences between the two conditions is needed, and some experiments are in progress.

Growth of G. pallidus H9 under aerobic and anaerobic conditions. a Effect of temperature. b Effect of salinity; the experiments were performed in LB medium at 65°C (200 rpm). Growth was determined by measuring optical density at 600 nm. Standard deviation was 0.1 ± 0.05
Degradation kinetics under aerobic and anaerobic conditions
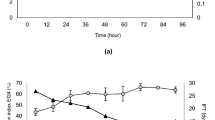
Figure 3 shows the typical time-course profile of bacterial biomass, biosurfactant production, CSL, emulsification index and surface tension under aerobic and anaerobic degradation conditions. The cell growth of aerobic culture reached its maximum high value at 4 days and then decreased from 11.2 g/L to 1.26 g/L. But the maximum biomass for anaerobic culture was reached after 36 days, and then decreased from 9.11 g/L to 6.1 g/L. This was possibly due to the use of different electron acceptors between the two conditions, with the oxidation–reduction potential of oxygen being higher than that of nitrate radical. The biosurfactant yield of the aerobic culture reached a maximum value (9.8 g/L) at 12 days and then decreased from 9.8 g/L to 4.29 g/L, while under anaerobic conditions, biosurfactant production increased slowly, reached a maximum (2.16 g/L) after 36 days, and then decreased from 2.16 g/L to 0.29 g/L. This indicated that biosurfactant synthesis is growth associated. The maximum production of biosurfactant under aerobic condition was more than that one observed under anaerobic condition, possible also due to the different electron acceptors that result in the different pathway of biodegradation which have different reaction activation energy and may produce different catabolic enzymes (Zengler et al. 1999; Röling et al. 2003; Todd Townsend et al. 2003; Siddique et al. 2006; Vasileva Tonkova and Gesheva 2007).

Degradation curve of strain H9. a Aerobic, b anaerobic. △ Cell surface hydrophobicity, ■ emulsification index 24 (EI24), X surface tension, ● biosurfactant (BS) yield, ◆ biomass. Standard deviation is 2.0 ± 0.5
An important property of biosurfactants is to reduce the surface tension of the medium. The surface tension reduction, CSL increase and emulsification potential increase paralleled cell growth. The surface tension of the medium decreased from 67.59 mN/m to 24.56 mN/m and 67.73 mN/m to 32.25 mN/m, respectively (Fig. 3). Meanwhile, experiments were done to evaluate CSL along with the process of the degradation. The results obtained from the BATH test showed that the CSL was changed with respect to the biosurfactant generated. The maximum value of CSL was 88.4% and 65% under the two degradation conditions, respectively. The presence of biosurfactant as connection between cell and oil was able to promote the attachment of the cell to the oil-soluble carbon source. However, the CSL of anaerobic degradation was lower than under aerobic conditions, possibly due to the different electron acceptor, and different composition and yield of the two conditions. Under aerobic conditions, oxygen was a more favorable electron-acceptor in the metabolic mechanism of strain H9, which has high oxidation-reduction potential and may promote the degradation of crude oil and the synthesis of biosurfactant. Under anaerobic conditions, nitrate is the main favorable electron-acceptor. The redox potential of nitrate is lower than that of oxygen and may generate a different oxidase and altered metabolic pathway, decreasing biosurfactant production.
In addition, the emulsification activity results showed the ability of strain H9 to emulsify immiscible hydrocarbon such as LHP crude oil. EI24 was maintained at 90–95% and 10–35% under aerobic and anaerobic conditions, respectively, and viscosity fell from ≈ 45.5 to ≈ 4.6 mPa·s (aerobic condition) s and from ≈ 45.5 to ≈ 24.2 mPa·s (anaerobic conditions), decreasing by about 89.89% and 46.81%, respectively. This indicated that some fraction of the crude oil had dissolved in water with biosurfactant, and formed a micro-emulsion. These results differ from those of Lotfabada et al. (2009), who found that Pseudomonas aeruginosa had the ability to produce biosurfactants and emulsify kerosene at 60.6% activity. Comparing these results with other biosurfactant-producing microorganisms (Margaritis et al. 1979; Lotfabada et al. 2009) suggests that strain H9 may be a good candidate for MEOR due to its different pattern of metabolism.
Analysis of biosurfactant
Color development reaction results showed a purplish-red spot on silica gel plates when using ninhydrin as a color developing reagent, suggesting that a peptide was found in this biosurfactant. Meanwhile, the appearance of a blue-green spot proved the presence of glycosides when using anthrone as color developing reagent, and the appearance of blue-black spot revealed the presence of lipids when using ammonium-perchlorate as color developing reagent. Accordingly, the biosurfactant excreted by strain H9 under both aerobic and anaerobic conditions contains three components: glycosides, peptide, and lipids. However, the quantities of these three components differ between the two conditions. Based on the phenol-sulfuric acid method, the biosurfactant synthesized under the two conditions contain (%, w/w): glycosides of ≈ 50.3 and ≈ 53.8 when using glucose as standard curve reagent, lipids of ≈ 35.4 and ≈ 31.2 when using dichloromethane-methanol method, peptide of ≈ 15.2 and ≈ 26.0 when using crystalline bovine serum albumin as standard curve reagent, respectively. This indicates that the biosurfactants synthesized by strain H9 were different under the two conditions, suggesting that their critical micelle concentration (CMC) is different.
Critical micelle concentration determination
The CMC can act as an indicator of the surface activity of a surfactant. Different surfactants have different CMCs. Surfactants with a lower value of CMC mean that the concentration for this surfactant to form a micelle is also lower.
The surface tensions of the purified biosurfactant solutions are presented in Fig. 4. This figure shows that the surface tension of biosurfactant decreased with increasing biosurfactant concentration until its lowest value was reached (aerobic condition: 25.09 mN/m and anaerobic condition: 30.15 mN/m). At this point, the concentration of biosurfactant, namely the CMC, was 16 mg/L for aerobic conditions and 22 mg/L for anaerobic conditions. The two biosurfactants exhibited different surface activities because of their different components; however, their CMCs were much lower compared with some chemical surfactants. For instance, sodium dodecyl sulfate (SDS) has a CMC value of 2,100 mg/L (Yin et al. 2009). Nitschke and Pastore reported that the CMC of biosurfactant obtained from Bacillus subtilis was 33 mg/L (Nitschke and Pastore 2006). Haba et al. (2000) showed that the CMC of rhamnolipid-degradation liquor was 386 mg/L.

The critical micelle concentration (CMC) of the biosurfactant produced by H9 under aerobic and anaerobic conditions was determined by the profile of the surface tension and concentration of biosurfactant. Two tangent lines of the curve converge upon one point, this point determines the CMC (0.016 g/L and 0.022 g/L, respectively). a Aerobic, b anaerobic. Standards deviation is 0.02 ± 0.005
Analysis of crude oil
It has been stated that using microbes for biodegrading crude oil may have the benefits of MEOR, such as sweeping oil, reducing oil viscosity and improving flow characteristics, altering wettability, minimizing paraffin precipitation or deposition problems along the production flow line, increasing API gravity value and finally reducing both the pour point and paraffin content of crude oil (Sadeghazad and Ghaemi 2003; Lazar et al. 1999). Gas chromatographic (GC) analysis of crude oil was carried out before and after treatment with strain H9 under aerobic or anaerobic conditions, and revealed marked changes.
Figure 5a illustrates the redistribution of saturated-hydrocarbons in LHP crude oil before and after biodegradation with strain H9 under aerobic conditions. There was a significant increase in the relative concentration of carbon fractions from C9 to C22 accompanied by a decrease in heavy fractions C23+ from 38.26 % to 35.27 % and short-chain fractions C8–C9 from 1.93% to 0.86%, respectively. The long-chain hydrocarbon fractions (C41, C42 and C43) were degraded completely and disappeared. Table 3 shows that the maximum peak of the relative concentration of carbon fraction was changed from C23 to C19, and the significant increase in the relative concentration of the carbon fractions ratio of w(C21−)/w(C22+), w(C21 + C22)/w(C28 + C29) accompanying with the decrease of w(pr)/w(nC17) and w(pr)/w(nC18). These results indicate that strain H9 has a preference for utilizing medium- and long-chain hydrocarbons under aerobic conditions.

Carbon concentrations before and after treatment with strain H9 under a aerobic and b anaerobic conditions. Standard deviation is 1.0 ± 0.2
A different trend was observed in the case of treatment with strain H9 under anaerobic conditions (Fig. 5b). The increase in the relative concentration of C12 to C31 from 86.27% to 90.1% is accompanied by a clearly significant reduction in the relative concentration of C8–C11 from 7.65% to 6.26% and C32+ from 6.08% to 3.64%, respectively. The long-chain hydrocarbons fractions (C39–C43) were degraded completely and disappeared. Table 3 shows that the maximum peak of the relative concentration of the carbon fraction changed from C23 to C20, and the carbon fractions ratio of w(C21−)/w(C22+), w(C21 + C22)/w(C28 + C29) was increased, accompanied by a decrease in w(pr)/w(nC17) and w(pr)/w(nC18). These results indicate that strain H9 has a preference for utilizing long chain hydrocarbons with a reduction in heavy hydrocarbon fractions (C32–C43) under anaerobic conditions. The evident difference in the metabolic pathways between aerobic and anaerobic degradation was possibly due to a change in redox potential during the biochemical reaction (Zengler et al. 1999; Röling et al. 2003; Todd Townsend et al. 2003; Siddique et al. 2006; Vasileva Tonkova and Gesheva 2007; Sousa et al. 2009).
However, under the two conditions, the biodegradation process carried out by strain H9 resulted in the same significant reduction of light hydrocarbon fractions (C8 and C9) and of heavy hydrocarbon fractions (C33 to C43), with an accompanying increase in hydrocarbon fractions (C12–C22). This may be due to the metabolism of long-chain alkane fractions compared to short-chain alkanes, or possibly to the fact that the low molecular weight hydrocarbons were volatilized easily and thus appear to be biodegraded. Similar results were also observed by Jobson et al. (1972) and Etoumi (2007). The results showed that saturated hydrocarbons increased greatly, accompanied by a reduction in aromatic hydrocarbons, non-hydrocarbon and asphaltene in crude oil under aerobic and anaerobic degradation over 100 days, and the oil degradation rate (%) of aerobic and anaerobic digestion was 58.5% and 44%, respectively (Table 4). As a consequence, GC analysis revealed that the physical-chemical characteristics of LHP crude oil had changed, becoming more mobile. This may be efficient in MEOR.
Viscosity and WAT
Figure 6 illustrates the changes in crude oil viscosity before and after microbial treatment under aerobic and anaerobic conditions with different shear rate values. Aerobic and anaerobic degradation had a clear impact; the viscosity of the crude oil decreased significantly from 45.5 Pa·s to 10.6 Pa·s under aerobic conditions, and from 45.5 Pa·s to 14.4 Pa·s under anaerobic conditions at 0.15 s−1. A sharp decrease in the viscosity at shear rates below 2 s−1 was observed, and the viscosity remained steady after 6 s−1. Comparing these results with other reports (Lazar et al. 1999; Sadeghazad and Ghaemi 2003; Hao et al. 2004) showed that strain H9 has the ability to degrade the heavy fraction and change the chemical and physical properties of LHP crude oil after both aerobic and anaerobic degradation.

Viscosity curves of crude oil treated with isolated species H9 under different conditions. Standard deviation is 1.0 ± 0.5
WAT was also used as an indicator of the microbially induced compositional changes of crude oil before and after microbial treatment. The results illustrated that WAT decreased significantly from 40.5°C to 37.5°C under aerobic conditions, while under anaerobic conditions, WAT was decreased similarly from 40.5°C to 38°C.
Oil recovery by the core flooding system
The results of core flooding experiments showed that the EOR values after first waterflooding were 61.06%, 61.49% and 59.77%, respectively. The MEOR was 7.46% for #1 core and 6.92% for #2 core. The oil recovery of #2 was lower than that of #1, possibly because #1 core consisted of two degrading patterns (aerobic and anaerobic biodegradation before and after oxygen was depleted completely during the incubation of 100 days) whereas #2 core consisted of one degrading pattern (anaerobic biodegradation).
Conclusion
The successful application of in situ MEOR depends largely on the capability of microorganisms to grow and produce metabolites under the extreme conditions found in oil reservoirs. Isolation and screening programs used here for LHP oil wells were based on the concept of obtaining bacterium that could produce biosurfactants. Geobacillus pallidus H9 was isolated and could grow at high temperature and salinity, and produce biosurfactant when using petroleum hydrocarbons as sole carbon source under both aerobic and anaerobic conditions. The results revealed certain significant differences between the pathways of metabolism under the two conditions. Core flooding system experiments showed that strain H9 and its biosurfactant have great potential for use in the oil industry for oil mobilization and MEOR, especially in high temperature and salinity oil reservoirs.
Reference
Afshar S, Lotfabad TB, Roostaazad R, Najafabadi AR, Noghabi KA (2008) Comparative approach for detection of biosurfactant producing bacteria isolated from Ahvaz petroleum excavation areas in south of Iran. Ann Microbiol 58:555–560
Al-Tahhan RA, Tahhan AL, Sandrin TR, Bodour AA, Maier RM (2000) Rhamnolipid-induced removal of lipopolysaccharide from Pseudomonas aeruginosa: effect on cell surface properties and interaction with hydrophobic substrates. Appl Environ Microbiol 66:3262–3268
Altschul SF, Gish W, Mille W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Brown LR (2010) Microbial enhanced oil recovery. Curr Opin Microbiol 13:316–320
Chayabutra C, Ju LW (2000) Degradation of n-hexadecane and its metabolites by Pseudomonas aeruginosa under microaerobic and anaerobic denitrifying conditions. Appl Environ Microbiol 66:493–498
Cooper DG, Goldenberg BG (1987) Surface-active agents from two Bacilllus species. Appl Environ Microbiol 53:224–229
Desai JD, Banat IM (1997) Microbial production of surfactants and their commercial potential. Microbiol Mol Biol Rev 61:47–64
Dubois M, Gills KA, Hamilton JK, Rebers PA, Smith F (1956) Colorimetric method for determination of sugar and related substances. Anal Chem 28:350–356
Etoumi A (2007) Microbial treatment of waxy crude oils for mitigation of wax precipitation. J Pet Sci Eng 55:111–121
Garrity GM, Brenner DJ, Krieg NR, Staley JT (2004) Taxonomic outline of the prokaryotes. Bergey’s manual of systematic bacteriology. May 2005, 2nd edn, vol 2. Springer, New York. DOI: 10.1007/bergeysoutline200405
Haba E, Espuny MJ, Busquets M, Manresa A (2000) Screening and production of rhamnolipids by Pseudomonas aeruginosa 47 T2 NCIB 40044 from waste frying oils. J Appl Microbiol 88:379–387
Han J, Duan WZ (1998) Feasibility study of extracting lipids by dichloromethane substituting for chloroform. Chinese J Anal Chem 26:1164–1167
Hao RX, Lu A, Zeng YS (2004) Effect on crude oil by thermophilic bacterium. J Pet Sci Technol Eng 43:247–258
Jenneman GE (1989) Potential of in situ microbial enhanced oil recovery. In: Donaldson EC, Chilingarian GV, Yen TF(eds) Microbial enhanced oil recovery, Chapter 3. Elsevier, Amsterdam, pp 37–69
Jobson A, Cook FD, Westlake DWS (1972) Microbial utilization of crude oil. J Appl Microbiol 23:1082–1089
Lazar I, Voicu A, Nicolescu C, Mucenica D, Dobrota S, Petrisor IG, Stefanescu M, Sandulescu L (1999) The use of naturally occurring selectively isolated bacteria for inhibiting paraffin deposition. J Pet Sci Technol Eng 22:161–169
Li HY, Zhang JJ (2003) Comparison of different methods on determining wax appearance temperature of crude oils. Oil Gas Storage Transport 22:28–30
Lotfabada TB, Shourianc M, Roostaazada R, Najafabadi AR, Adelzadeh MR, Noghabi KA (2009) An efficient biosurfactant-producing bacterium Pseudomonas aeruginosa MR01, isolated from oil excavation areas in south of Iran. Colloids Surf: B 9:183–193
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Manocha MS, San-Blas G, Centeno S (1980) Lipid composition of Paracoccidioides brasilienses: possible correlation with virulence of different strains. J Med Microbiol 117:147–154
Margaritis A, Zajic JE, Gerson DF (1979) Production and surface active properties of microbial surfactants. Biotechnol Bioeng 21:1151–1162
McInerney MJ, Sublette KL (2002) Oil field microbiology. In: Hurst CJ, Stetzenbach D (eds) Manual of environmental microbiology, 2nd edn. ASM, Washington DC, pp 777–778
Miller GL, Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes. Appl Environ Microbiol 27:985–987
Nitschke M, Pastore GM (2006) Production and properties of a surfactant obtained from Bacillus subtilis grown on cassava wastewater. Bioresour Technol 97:336–341
Pruthi V, Cameotra SS (2003) Effect of nutrients on optimal production of biosurfactants by Pseudomonas putida-a Gujarat oil field isolate. J Surfactants Deterg 6(01):65–68
Reddy CM, Quinn JG (1997) Environmental chemistry of benzothiazoles derived from rubber.Environ Sci Technol 31:2847–2853
Robert M, Mercade ME, Bosch MP, Parra JL, Espuny MJ, Manresa MA, Guinea J (1989) Effect of the carbon source on biosurfactant production by Pseudomonas aerugonosa 44Ti. Biotechnol Lett 11(12):871–874
Rojo F (2009) Degradation of alkanes by bacteria. Environ Microbiol 11(10):2477–2490
Röling WFM, Head IM, Larter SR (2003) The microbiology of hydrocarbon degradation in subsurface petroleum reservoirs: perspectives and prospects. Res Microbiol 154(05):321–328
Rosenberg M (1984) Bacterial adherence to hydrocarbons: a useful technique for studying cell surface hydrophobicity. FEMS Microbiol Lett 22(03):289–295
Sadeghazad A, Ghaemi N (2003) Microbial prevention of wax precipitation in crude oil by biodegradation mechanism. Soc Pet Eng SPE 80529:1–11
Salminen JM, Tuomi PM, Suortti A-M, Jørgensen KS (2004) Potential for aerobic and anaerobic biodegradation of petroleum hydrocarbons in boreal subsurface. Biodegradation 15:29–39
Sanchez G, Marin A, Vierma L (1992) Isolation of thermophilic bacteria from a Venezuelan oil field. In: Premuzic ET, Woodhead A (eds) Microbial enhancement of oil recovery—recent advances. Proceedings of the (1992) International Conference. Elsevier, Amsterdam, pp 383–389
Siddique T, Fedorak PM, Foght JM (2006) Biodegradation of short-chain n-alkanes in oil sands tailings under methanogenic conditions. Environ Sci Technol 40(17):5459–5464
Sousa DZ, Smidt H, Alves MM, Stams AJM (2009) Ecophysiology of syntrophic communities that degrade saturated and unsaturated long-chain fatty acids. FEMS Microbiol Ecol 68(03):257–272
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary geneticsanalysis (MEGA) software version 4.0. Mol Biol Evol 24(08):1596–1599
Todd Townsend G, Prince RC, Suflita JM (2003) Anaerobic oxidation of crude oil hydrocarbons by the resident microorganisms of a contaminated anoxic aquifer. Environ Sci Technol 37(22):5213–5218
Vasileva Tonkova E, Gesheva V (2007) Biosurfactant production by antarctic facultative anaerobe Pantoea sp. during growth on hydrocarbons. Curr Microbiol 54:136–141
Winker S, Woese CR (1991) A definition of the domain Archaea, Bacteria and Eucarya in terms of small subunit ribosomal RNA characteristics. Syst Appl Microbiol 14:161–165
Yakimov MM, Timmis KN, Wray V, Fredrickson HL (1995) Characterization of a new lipopeptide surfactant produced by thermotolerant and halotolerant subsurface Bacillus licheniformis BAS50. Appl Environ Microbiol 61:1706–1713
Yan F, Xie YJ (2009) Investigation on separation of four fractions and interfacial properties of DaQing crude oil. Chem Anal Meterage 18:20–24
Yin H, Qiang J, Jia Y, Ye JS, Peng H, Qin HM, Zhang N, He BY (2009) Characteristics of biosurfactant produced by Pseudomonas aeruginosa S6 isolated from oilcontaining wastewater. Process Biochem 44:324–331
Youssef N, Simposn DR, Duncan KE, McInerney MJ, Folmsbee M, Fincher T, Knapp RM (2007) In situ biosurfactant production by Bacillus strains injected into a limestone petroleum reservoir. Appl Environ Microbiol 73:1239–1247
Zengler K, Richnow HH, Michaelis W, Rosselló-Mora R, Widdel F (1999) Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401:266–269
Acknowledgments
This manuscript was supported financially by the “National High Technology Research and Development Program of China, grant: 2009AA063504”, “the National Basic Research Program of China, grant 2005cb221308” and “the Science research and technology Exploitation program of PetroChina Company Limited, grant: 2008A-1403” for microorganism and metabolite to enhance oil recovery.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wenjie, X., Li, Y., Ping, W. et al. Characterization of a thermophilic and halotolerant Geobacillus pallidus H9 and its application in microbial enhanced oil recovery (MEOR). Ann Microbiol 62, 1779–1789 (2012). https://doi.org/10.1007/s13213-012-0436-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-012-0436-5