
Abstract
Colletotrichum falcatum, an ascomycete pathogen causes red rot of sugarcane which is specialized to infect cane stalks. Cellulolytic and pectinolytic enzymes are necessary for degradation of plant cell wall which stands as barrier for successful fungal pathogenesis. In the study, we have confined to the CAZy genes that regulate cellulolytic and pectinolytic enzymes in two distinctive pathotypes of C. falcatum. Comparative transcriptome analysis revealed that a number of CAZy genes producing cellulolytic and pectinolytic enzyme were present in the virulent (Cf671) and least virulent (RoC) pathotypes. Two consecutive transcriptome analyses (in vitro) were performed using Illumina Hi Seq 2500, further analysis was done with various bioinformatic tools. In vitro expression analysis of cutinase, glycoside hydrolyase and pectin-related genes revealed number of genes that attributes virulence. Numerous pectin-related genes involved in degradation of plant cell wall, pectinase and pectin lyase are considered to be key precursor in degradation of pectin in sugarcane. These results suggest that cellulolytic enzymes, cutinase and pectin-related genes are essential for degradation of sugarcane cell wall and considered to be an important pathogenic factor in C. falcatum. This is the first detailed report on sugarcane cell wall-degrading enzymes during its interaction with C. falcatum and also this comparative transcriptome analysis provided more insights into pathogen mechanism on C. falcatum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The aerial parts of plants have outer layer which is considered to be first line of defence against intervention of plant pathogens; cuticle is the first challenging factor for the pathogen during its invasion (Chen et al. 2013). Fungal pathogens have an ability to produce arsenal of proteins and enzymes that can overcome and interfere pathogen-associated molecular pattern (PAMP) triggered immunity (PTI) and effector triggered immunity (ETI) (Jones and Dangl 2006). The serine esterase made of α/β hydrolase superfamily serves as cutinase will facilitate the pathogen invasion into the host system by hydrolysing the cuticle layer with the help of small monomers of cutin (Lu et al. 2018; Nikolaivits et al. 2018). There are several evidential reports suggesting that cutinases are involved in spore attachment, appressorium formation, initial attachment/degradation of plant cutin layer in various ascomycetes, and still there exists catalogue of confusion in determining the role of cutinase (Lu et al. 2018; Deising et al. 1992; Stahl and Schafer 1992; Koschorreck et al. 2010; Zhang et al. 2014).Though there are plenty of reports suggesting that cutinase has various roles in host pathogen interaction, the role of cutinase in sugarcane during interaction with C. falcatum has not been exploited. The damage associated molecular pattern (DAMP) which is considered to be rapid in triggering innate response than PAMP triggered response, helps plant cell wall reinforcement, biosynthesis of antimicrobial compounds and phytoalexins. Pectin is considered to be one of the major components in plant cell wall, especially in between the cells/xylem vessels and in woody tissues, which acts as secondary cell wall. The role of pectin lyase, polygalacturonase in sugarcane serves as inducer in producing high biomass yield and this pectin is composed of acidic and neutral sugar-containing side chains which is utilised highly as renewable biomass co-product (Xiao and Anderson 2013). Pectin layer in sugarcane is considered to be most important primary cell wall component which comprises 35% approximately in dicot and several monocots (Lara-Marquez et al. 2011).
Colletotrichum falcatum Went (Perfect state; Glomerella tucumanensis (Speg.) Arx & Muller) infects the cane stalks and causes symptoms of rotting of internodal tissues, inversion of sucrose by producing of pathogen induced invertases and death of cane stalks. Numerous references were recorded on economic importance in many sugarcane growing countries over early years of the past century (Viswanathan 2010, 2021a). The disease remains a major constraint to sugarcane production in India, Pakistan, Bangladesh, Indonesia, Myanmar, Thailand, Vietnam, USA, Sudan, etc. (Singh et al. 2000; Viswanathan 2021a). Epidemics of the disease have been very common ever since its occurrence in India in 1901 which spread from Saccharum officinarum/ S. barberi clones to the newly released varieties developed through nobilization (Viswanathan 2021b). Further, the cane cultivation and sugar industries in the past century have witnessed lots of high-yielding sugarcane varieties being uprooted from the field due to red rot. Although the disease is effectively managed by releasing disease-resistant varieties (Viswanathan and Samiyappan 1999; Viswanathan and Rao 2011), the resistance in the varieties is not static due to emergence of new pathogenic strains that cause breakdown of resistance in the popular sugarcane varieties (Viswanathan 2021b). C. falcatum exhibits enormous variation for pathogenicity and such extensive pathogenic variation has been documented by comparing virulence pattern in 117 C. falcatum pathotypes/ isolates under tropical and subtropical conditions (Viswanathan et al. 2017). The same study reported pathogenicity/ virulence of the designated pathotypes CF01, -02, -03, -04, -05, -06, -07, -08, -09, -10, -11 and -12 that destroyed the cvs Co 1148, Co 7717, CoJ 64, CoS 767, Co 419, Co 997, CoC 671, Co 94012 and 89V74 and also of other isolates and sugarcane varieties.
Further, the mechanism governing red rot resistance has not been fully understood due to genomic complexity. The differential induction of chitinases and thaumatin-like proteins (TLP’s) in sugarcane in response to infection by C. falcatum has been demonstrated using biochemical and Western blot studies (Viswanathan et al. 2005). Differential expression of genes during interaction of sugarcane x C. falcatum was established through DD RT-PCR assays (Pratima et al. 2013; Rahul et al. 2016). 2D based core proteome analysis classification was performed during sugarcane challenged with C. falcatum (Ganesh Kumar et al. 2020). Suppression subtractive hybridization (SSH) based time specific and initial defense responses of sugarcane during C. falcatum pathogenesis were recorded (Sathyabhama et al. 2015, 2016). However, the molecular tools have yet to be fully explored to characterize the genes activated during pathogen invasion. In this new-generation sequencing era, it is easy to exploit the genes responsible for an action using Next-generation sequencing (NGS) technology. Especially RNA-Seq technology will help us to explore the genes and their expressions to a great extent with their relative expression level. We have sequenced the whole genome and transcriptome of C. falcatum using Illumina Hi-Seq 2500 (Viswanathan et al. 2016; Prasanth et al. 2017). C. falcatum was found to have large number of plant cell wall-degrading enzymes (PCWDE) such as CAZy, secondary metabolites, cutinase and secretory effectors proteins. Earlier we reported candidate secretory effector proteins from genome and transcriptome of C. falcatum and virulent strain-specific SSR marker from the genome of C. falcatum (Prasanth et al. 2019, 2021). In that, CAZy profusely found to play a vital role in cell wall degradation, host target identification, biotrophy–necrotrophy switch, etc. In the present study, a comparative transcriptome analysis of cellulolytic and pectinolytic enzymes in two pathotypes Cf671 (virulent) and CfRoC (least virulent) of C. falcatum has brought a revolutionary identification a large number of CAZy genes contributing to plant cell wall degradation, regulating cellulolytic enzymes and pathogenic virulence.
Materials and methods
Fungal culture used
The C. falcatum pathotype CfRoC (isolated from the sugarcane variety RoC) maintained at the red rot culture collection of the institute (ICAR-Sugarcane Breeding Institute, Coimbatore India) was used for the current study. The fungus was multiplied on oatmeal agar (oatmeal 30 g, agar 20 g, water lL) for 7 days. Monoconidial culture of the fungus was inoculated on complete media broth (CMB) and incubated under room temperature (28 ± 2 °C) for 12 days (without shaking) for RNA isolation. Previously sequenced C. falcatum pathotype Cf 671 (isolated from the sugarcane variety CoC 671) was taken for comparative transcriptome analysis (Prasanth et al. 2017).
RNA extraction and library preparation for transcriptome sequencing
Total RNA was extracted from C. falcatum CfRoC using TRI Reagent (Sigma-Aldrich, USA) and treated with Rnase free DNAse I (Promega, USA). Subsequently, the quality of RNA was checked in 1% denatured agarose gel electrophoresis for the presence of intact 28 and 18S bands and RNA was quantified using Nanodrop-8000. The paired-end cDNA sequencing library was prepared using Illumina TruSeq SBS Kit v3 as per the manufacture’s protocol. Library preparation was started with mRNA fragmentation followed by reverse transcription, second-strand synthesis, paired-end adapter ligation and finally ended with index PCR amplification of adaptor-ligated library. Library quantification and quality check were performed on Caliper Lab Chip GX using HT DNA High Sensitivity Assay Kit. The libraries of all samples were in the size range of 200 bp to 600 bp. The resulting libraries were validated using the Agilent Bio Analyzer 2100 onto the Agilent High Sensitivity Chip performed at Nucleome Informatics Pvt. Ltd, Hyderabad, India.
Cluster generation and sequencing run
Paired-End sequencing allows the template fragments to be sequenced in both forward and reverse directions. Cluster generation was carried out by hybridization of template molecules onto the oligonucleotide-coated surface of the flow cell. Immobilized template copies were amplified by bridge amplification to generate clonal clusters. This process of cluster generation was performed on cBOT using TruSeq PE Cluster kit v3-cBot-HS. The kit reagents were used for binding of samples to complementary adapter oligos on paired-end flow cells. The adapters were designed to allow selective cleavage of the forward cDNA strand after resynthesis of the reverse strand during sequencing. The copied reverse strand was then used to sequence from the opposite end of the fragment. TruSeq SBS v3-HS kits were used to sequence cDNA of each cluster on a flow cell using sequencing by synthesis technology on the HiSeq 2500.
Transcriptome sequencing, assembly and annotation
The cDNA libraries prepared and sequenced on the Illumina HiSeq 2500 and both ends of the sequenced cDNA were analysed using FastQC v0.11.3. Clean reads were obtained by removing the empty reads, the adaptor sequences and the low-quality sequences (reads with unknown base airs ‘N’) using TRIMMOMATIC V 0.33. The clean reads were then assembled into contigs and transcripts based on pair-end information using CLC genomics workbench V 6.0 on default parameters. The CDS regions were predicted using Trans Decoder V 2.0.1. The C. falcatum transcriptome was annotated using BLASTX and BLAST2GO. The KEGG pathway annotation was performed using KAAS automated server.
Gene enrichment analysis
GO sequence distribution, helped in specifying all the annotated nodes comprising of GO functional groups. CDS of transcripts associated with similar functions were assigned to the same GO functional group. The GO sequence distributions were analysed for all the three GO domains, i.e. biological processes, molecular functions and cellular components.
KEGG pathway identification
Orthologous assignment and mapping of the transcripts to the biological pathways were performed using KEGG automatic annotation server (KAAS) (Moriya et al. 2007). All the transcript contigs were compared against the KEGG database (Kanehisa and Goto 2000; Kanehisa et al. 2012) using BLASTx with threshold bit-score value of 60. The mapped transcript contigs represented metabolic pathways of major biomolecules such as carbohydrates, lipids, nucleotides, amino acids, glycans, cofactors, vitamins, terpenoids, polyketides, etc. The mapped contigs also represented the genes involved in genetic and environmental information processing and cellular processes.
Screening protein families involved in fungal pathogenesis
Protein families were classified by searching the assembled transcripts against Pfam (Punta et al. 2012) and InterProScan (Quevillon et al. 2005). Protease families were identified using BLASTp against MEROPS peptidase database release 9.6 (Rawlings et al. 2012). Cytochromes (CYPs) were named according to classification details collected from BLASTp against Fungal Cytochrome P450 database version 1.2 (Park et al. 2008). Carbohydrate-degrading enzymes selected from InterProScan and Pfam analyses were classified according to GH (Glycoside hydrolase) family as classified in CAZy database (Cantarel et al. 2009). Proteases, carbohydrate-degrading enzymes and membrane transporters were predicted from C. falcatum transcriptome and compared with C. graminicola, C. sublineola C. higginsianum, C. gloeosporioides and C. orbiculare.
RT-PCR validation
To validate the candidate genes selected from each family of CAZy based on the transcriptome data, cell wall glycosyl hydrolase, glycoside hydrolase family 5, glycoside hydrolase family 3, glycoside hydrolase family 10, glycoside hydrolase family 35, glycoside hydrolase family 43, glycoside hydrolase family 28, glycosyl transferase, polysaccharide deacetylase, polysaccharide deacetylase 3 and carbohydrate esterase by quantitative reverse transcription polymerase chain reaction (qRT-PCR). The assay was performed with a Step-One plus Real-Time PCR system (Applied Bio systems, USA) to profile the expression of cellulolytic, hemi cellulolytic and pectinolytic genes which are involved in plant cell wall degradation in virulent (Cf671) and avirulent (CfRoC) C. falcatum pathotypes. The CAZys retrieved from comparative transcriptomics gave a clear view about up regulation and down regulation between the pathotypes of C. falcatum. Each reaction was carried out in triplicate; the primers used for qRT-PCR were designed based on the transcriptome data except for actin and are listed in Table ESM 4. The actin primers described by Chakravarthi et al. (2015) were used for normalization. For qRT-PCR experiments, cDNA concentration was standardized for each sample and dissociation curve analysis was performed to check primer specificity. The cocktail for qRT-PCR reaction contained 50 ng cDNA, 2.5 pmol primers, 10 µl SYBRGREEN Master Mix, and nuclease free water to make up the reaction mixture to 20 µl. The reaction was performed for 40 cycles (denaturation for 10 min at 95 °C followed by annealing and extension at 1 min for 58 °C).
Results
Culture morphology and transcriptome assembly
Cf671 and CfRoC pathotypes were morphologically distinct and they have their uniqueness in their survival and development. The histopathological study on C. falcatum pathotypes brought a clear outline that virulent pathotype had ability to produce orange/salmon coloured acervuli whereas CfRoC was non-sporulating did not produce any visible conidia (Figs. 1 and 2). The pathotype Cf671 generated 56, 637,987 reads and the pathotype CfRoC generated 55,664,294 reads (Table ESM 1). The trimmed high-quality reads were assembled using TRINITY v2.0.6; the short reads passed QC were assembled into transcripts. The contig length of Cf671 pathotype was 31,194,966, whereas in case of CfRoC pathotype it was 33,092,606 (Table ESM 1).

Cultures of C. falcatum sporulating and non sporulating pathotypes on oat meal agar; (A) sporulating highly virulent pathotype Cf671. (B) Non sporulating and less virulent pathotype CfRoC

Phenotypic disease symptoms of C. falcatum after plug and nodal swab inoculation in sugarcane stalks of susceptible cv CoC 671. A Cf671 pathotype inoculated; B CfRoC pathotype inoculated. Arrows indicate site of pathogen inoculation in the plug method and circles indicate the pathogen inoculated node in nodal swabbing method. Only in the pathotype Cf671 inoculated canes disease progress is noticed in both the methods
Annotations
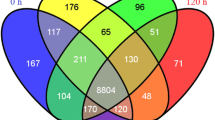
The transcriptome of Cf671 pathotype was assembled into 24,732 transcripts and 30,510 transcripts in case of CfRoC pathotype (Table ESM 2). The assembled transcripts were further annotated using CD-HIT-EST v4.6.1 (12), TGICL v2.1 (13), TransDecoder v2.0.1, BlastX and Blast2GO. This TransDecoder revealed that there were 23,136 CDS regions in the pathotype Cf671 and 17,830 CDS regions in CfRoC. The transcriptome of the pathotype Cf671 was further annotated, based on BlastX search in that 19,183 were found to be annotated while 5549 had no significant blast hits among the 24,732 transcripts. About 21,221 were found to be annotated and 9289 unigenes had no significant blast hits in case of CfRoC. The species distribution for the Cf671 using BlastX revealed that 9046 genes have best matches (first hit) with C. graminicola, followed by C. sublineola (8125), C. higginsianum (1158), C. gloeosporioides (465), C. orbiculare (295), Nectria haematococca (161), Verticillium albo-atrum (77) Magnaporthe oryzae (41), Chaetomium globosum (41), Metarhizium acridum (28), Neurospora crassa (16), other fungal organisms (296), other organisms (845) (Fig. 3). The annotations from CfRoC revealed that the C. graminicola has 9243 common genes between them, followed by C. sublineola (8448), C. higginsianum (1206), C. gloeosporioides (568), C. orbiculare (182), C. fioriniae (629), other fungal organisms (694)*, and other organisms (199) (Fig. 3). Even though the genes sharing between Colletotrichum species are closer, they do have their own novel genes which make differences among survival in their hosts system. The total number of unique annotated proteins from the pathotype Cf671 was 4356 and 12,142 proteins from the pathotype CfRoC. The common annotated transcripts between the pathotypes Cf671 and Cf RoC were found to be 5251 and 8169, respectively.

Hierarchical gene cluster analysis and common fungal transcript annotation statistics based on organisms; the majority of gene sharing is between C. graminicola and C. sublineola in both the pathotypes
Gene family’s expansion and contraction
The gene family’s expansion and contraction in the fungal pathogen are associated with the particular host range. In case of C. falcatum, it is more specific and the pathotype Cf671 which is considered to be virulent has contracted several classes of genes for colonization and establishing virulence in the host system, whereas the least virulent pathotype Cf RoC had a greater number of genes than the virulent pathotype. The major group of genes which contributes virulence in a hemibiotrophic pathogens CSEPs, Secondary Metabolites, Transporters and CAZy, are found to be contracted in the pathotype Cf671 and expanded in the pathotype CfRoC (Table ESM 3).
GO and KEGG distribution
The GO sequence distributions for Cf671 and CfRoC pathotype were analysed for all the three GO domains, i.e., biological processes, molecular functions and cellular components. The Cf671 had 8098 genes with 23 functional groups, whereas the pathotype CfRoC had 23,910 genes distributed from transcripts (Fig. 4). A total of 2399 unigenes were predicted as pathway regulatory genes and were annotated in the KAAS database, assigned to several KEGG pathways in Cf671; however, 5547 unigenes were found to be present and predicted several regulatory pathway genes in CfRoC.

KEGG distribution between C. falcatum pathotypes of Cf671 and CfRoC
CAZy distribution
Carbohydrate active enzymes (CAZy) play a vital role in degradation of plant cuticle layer by triggering the fungal plant cell wall-degrading enzymes (PCWDEs). C. falcatum has plenty of PCWDEs which belong majorly to glycoside hydrolyase (GH) and also CAZy contributes to biotrophy–necrotrophy switch. The major classes of CAZy glycoside hydrolyase (GH), glycosyl transferase (GT), polysaccharide lyase (PL), carbohydrate esterase (CE) and carbohydrate-binding modules (CBM) are found to be present in C. falcatum (Fig. 5). The heat map and distribution of CAZy family among the pathotypes of C. falcatum brought a clear view that they are mainly involved in cellulose degradation and pathogenicity development in Cf671 and Cf RoC and also for survival in the host system, respectively (Fig. 7).

Different classes of CAZy families and its distribution in C. falcatum pathotypes Cf671 and CfRoC transcriptome analyses
Enzymes involved in plant cell wall degradation
Most of hemibiotrophic fungal organism secrete cell wall-degrading enzymes in large numbers, among them cellulose, hemicellulose and pectin-degrading enzymes play a vital role. Transcriptome analysis revealed presence of several enzymes likes xylanases, cutinases, cellobiohydrolyase, alpha-glucosidase, alpha rhamnosidase, pectate lyase, galactomannan, alpha-mannosidase and ligninolytic enzymes like laccases and peroxidases in C. falcatum.
Functional analysis of CAZy in C. falcatum
qRT-PCR assay was performed for the candidate genes selected from each family of CAZy to profile the expression of cellulolytic, hemicellulolytic and pectinolytic genes in C. falcatum. The CAZys retrieved from comparative transcriptomics gave a clear view about up regulation and down regulation between the pathotypes of C. falcatum. The qRT-PCR profiles showed that the candidate genes are considered to be putative pathogenicity genes which are involved in virulence losses/gain. Candidate genes for cellulose such as cell wall glycosyl hydrolase, glycosyl hydrolase 3 and 5 were up regulated in both the pathotypes Cf671 and CfRoC at in vitro stage. During in planta interaction, glycosyl hydrolase 10 (xylanases) and glycosyl hydrolase 35 (β-galactosidases) from hemicellulose and polysaccharide deacetylase (pectin degrading) were upregulated in the pathotypes Cf671 but downregulated in CfRoC (Fig. 8). In contrast, the up regulation and down regulation between the Cf671 and CfRoC enzymes such as GH, GT and PL may probably regulate the pathogenicity in C. falcatum.
Discussion
Plant cell wall is the major barrier to the establishment of fungal infection on a host system. Cellulose, the major polysaccharide is present in the plant cell wall; therefore, fungal organism produces specific enzymes to degrade rigid wall and to surpass the layer of protection by hydrolysing cellulose and its derivatives (Laine et al. 2000; Wanjiru et al. 2002). The pathogen invasion and biotrophic development mask its surface by converting the hyphal surface-exposed chitin by deacetylation. This chitin degrades plant cuticle layer and elicits defense responses (Gueddari et al. 2002). Most of the Colletotrichum spp. produces a number of cell wall-degrading enzymes both in in planta and in vitro conditions (Gan et al. 2013; Prasanth et al. 2017). There are several reports in Colletotrichum that PCWDEs have been implicated in facilitating successful invasion and colonization in the host system (Choi et al. 2013; Rao and Nandineni 2017; O’Connell et al. 2012; Gan et al. 2013). The arsenal of C. falcatum genes involved in hemibiotrophic lifestyle is found to be similar to C. graminicola and C. sublineola infecting related hosts maize and sorghum, respectively (Viswanathan et al. 2016). The genome of C. falcatum revealed presence of PCWDEs, candidate secretory effectors (CSEPs), transposable elements, primary and secondary metabolites, membrane transporters, signalling molecules, CAZy and mating proteins involved in development and sclerotic management proteins. Though PCWDE are essential for successful pathogenesis and regulation of virulence in C. falcatum, their role during the process of pathogenesis is poorly understood. To exploit the ability to produce PCWDE in C. falcatum a comparative transcriptome approach has been performed with NGS platform using Illumina Hi Seq 2500. Two different pathotypes of C. falcatum varying in virulence were sequenced and their transcriptome analyses of gene expression profiles between Cf671 and CfRoC revealed that virulent pathotype produces a greater number of PCWDE genes when compared with other hemibiotrophs (Fig. 5). Most of the enzymes screened were produced in in vitro conditions; PCWDE enzymes in C. falcatum are capable enough to degrade pectin cellulose, and polysaccharides though cell wall possess a greater number of polysaccharides for their defence (Prasanth et al. 2017, 2019). This study aimed to advance the understanding of the PCWDEs such as cellulolytic, hemi cellulolytic and pectin-related genes present in C. falcatum causing red rot in sugarcane. As described above, this plant pathogenic fungus has large number of CAZy genes which are putatively involved in pathogenicity and they were characterized using virulent pathotype (Cf671) and a least virulent pathotype (CfRoC). In this study, we conducted a transcriptome sequencing of least virulent pathotype (Cf RoC) and compared it with virulent pathotype (Cf671) which was sequenced using NGS platform. Comparative transcriptomics brought an understanding that distribution of CAZy genes in each pathotype regulates their pathogenic mechanism which helps in their survival in the host system and also to be the successor in causing red rot in sugarcane.
Sugarcane stalk contains more of cellulose, pectin, cutin and wax that favour hard rigid cell wall as first barrier to fungal invasion (Viswanathan 2010). For pathogenic virulence in C. falcatum, it requires biochemical weapons to breach cell wall of sugarcane. Most of CAZy genes involved during appressorial phase in Colletotrichum spp, for example 18 different families, were reported in C. graminicola and 35 different CAZyme families in C. higginsianum (O’Connell et al. 2012). The role of CAZy is to potentially degrade polymers of plant cell wall such as cutin, cellulose, hemicelluloses and pectin. CAZyme regulates genes which are involved in initial penetration of host epidermal cell wall; large number of genes induced both in biotrophic and necrotrophic phases of Colletotrichum (O’Connell et al. 2012; Prasanth et al. 2017; Gan et al. 2012). Regulation of cellulolytic, hemi cellulolytic and pectin degradation by CAZy makes the pathogen to become great potential and induces virulence. The CAZy genes such as GH, GT, PL, CBM and CE gene families between Cf671 and CfRoC have been extensively studied using comparative transcriptome approach which revealed that the fungal pathogen C. falcatum establishes in the host tissues via fungal host-induced virulence effectors. Enzymes that encode modifications in fungal cell wall are glycoprotein synthesis/processing (GH47, GH76, and GH92), chitin deacetylases (CE4), chitinases and β-1,3-glucans (GH16, GH17, GH55, GH64) which were found to be present in C. falcatum. β-1,3-glucanases (GH16) induced in C. graminicola appressoria and involved in mechanical penetration were found to be present in both the pathotypes of C. falcatum (Fig. 6). Hemicellulase and pectinase activities and expanded class of CWDEs were reported in C. truncatum. The genes containing CBM42 binding to arabinofuranose were found in all fungi except in C. truncatum and C. incanum (Rao and Nandineni 2017). Analysis of major polysaccharide-degrading enzymes with wide range of substrates revealed linkage-specific expansion in glycosyl hydrolyase family. Our previous report on screening the effectors from the C. falcatum genome and transcriptome revealed that cell wall-degrading glycosyl hydrolyase was found to be highly induced at biotrophic stage (Prasanth et al. 2019). The fruit rot causing pectin-degrading enzymes such as GH 28, GH 78, PL 1 and PL 3 are found to be expanded in C. truncatum and C. fruticola, but in case of C. falcatum the number of genes was found to be on par but their roles vary as stage-specific genes. The contraction of auxiliary activity (AA) enzymes reported in C. graminicola and C. sublineola was found to be same in C. falcatum. The Heat map of cellulolytic, hemicellulolytic and pectin-degrading enzymes showed variation in their expression patterns between two pathotypes of C. falcatum (Fig. 7) which clearly shows that CAZy regulates cell wall-degrading enzymes and may play a vital role in plant penetration and successful pathogenesis (Fig. 8).

Distribution of cellulolytic, hemicellulolytic and pectin-related genes compared between C. graminicola and C. falcatum pathotypes Cf671 and CfRoC. The genes in Cf671 are up regulated compared to C. graminicola and CfRoC

Heat map for the expressed cellulolytic, hemicellulolytic and pectin degrading enzymes from two pathogenic pathotypes Cf671 and CfRoC. Glycoside hydrolase are found to be expressed higher in virulent pathotype, whereas glycosyl transferase expressed higher in the least virulent pathotype CfRoC

Transcriptome profiling of CAZy regulatory candidate genes based on their RPKM values, A-Cf cell wall glycosyl hydrolase; B-Cf glycosyl hydrolase 5; C-Cf glycosyl hydrolase 3; D-Cf glycosyl hydrolase 10; E-Cf glycosyl hydrolase 35; F-Cf glycosyl hydrolase 43; G-Cf glycosyl hydrolase 28; H-Cf polysaccharide deacetylase; I-Cf polysaccharide deacetylase 3
Cutinase plays a vital role not only in breakdown of leaf cuticle layers during penetration but also for degradation of plant cell wall during colonization and hyphal invasion. But in case of C. falcatum, a stalk-infecting pathogen, there are numerous cutinase genes such as glycosyl hydrolase and their roles vary depending on the developmental stages of the host. It is considered to be stage-specific expressive gene; their enzymes are involved in colonization and hyphal development in the host system. Pectate lyase in C. gloeosporioides promotes pathogenicity; to test their contribution in penetration and colonization in avocado fruit, a heterologous expression study was carried out and it was found that complexity of system requires more than one factor for pathogenicity (Yakoby et al. 2000). C. falcatum has a greater number of pectate lyase genes which may be putatively involved in pathogenesis and virulence factors. Based on stalk infecting nature, the pectin lyase sequences are different compared to other hemibiotrophs. The evolutionary effect of enzyme diversity is due to the substrates that are encountered during interactions with an extended variety of hosts. The Colletotrichum spp. have a wide range of hosts and close relationship. During the developmental stages they bypass plant defense and penetrate cell walls with the use of lytic enzymes. Host plants also have strategic approaches to detect and to defend against attack of pathogens by producing phytoalexins and inhibitor enzymes (Viswanathan and Rao 2011). Hemibiotrophic lifestyle in Colletotrichum spp. makes greater variability of unique enzymes based on the host system and most of them are regulated by CAZy genes. The relationships of lytic enzymes and the lifestyle of fungi are very well exploited after the advent of next generation sequencing technology, this high throughput sequencing method made several evidential reports in Colletotrichum spp that reveals about plant cell wall-degrading enzymes and role during pathogenesis. It is very well demonstrated that pathogenic fungi of monocotyledonous plants are better adapted to degrade the cell wall of plants reflecting host preference (Choi et al. 2013). Comparative transcriptome analysis of C. falcatum pathotypes reveals that there are three major PCWDEs such as cellulolytic, hemi cellulolytic and pectin-degrading enzymes which are regulated by CAZymes. Those enzymes were compared with other hemibiotrophs based on the following classification: The cellulolytic enzymes were classified as cellulases (GH6, GH7, GH5, GH12, and GH45), β-glycosidases (GH1 and GH3) and accessory enzymes such as GH 61 and CBM (Table 1) and the hemi cellulolytic enzymes as β-glucuronidases (GH67, GH115), xylanases (GH10, GH11, GH30), xyloglucanases (GH74), α-galacatosidases (GH27, GH36), β-mannase (GH26), α-arabinosidases (GH43, GH51, GH54) and β-galacatosidases (GH62, GH35) (Table 2). The pectinolytic enzymes such as polygalacturonases (GH28, GH78) and polygalacturonate lyase (PL1, PL3, PL4, PL9 and PL11) (Table 3) are found to be present. The obtained results suggested that C. falcatum pathotypes produce cellulolytic and pectinolytic enzymes in large numbers that helps to degrade sugarcane cell wall and involvement of these enzymes in development of red rot in sugarcane. The CAZy from Cf671 pathotype were found to be abundant than CfRoC pathotype and this is the first investigation on pathotype variation, genetic mechanism and simulation of pathogenicity between the C. falcatum pathotypes.
Data access
All data contributing to this transcriptome initiative have been deposited at the NCBI under BioProject PRJNA272832 and PRJNA380334. The accession number of Sequence Read Achieves (SRA) is SRR1765657. The Bio sample accession number of CfRoC is SAMN06640823 and SRA: SRS2076621.
Abbreviations
- CAZy:
-
Carbohydrate active enzymes
- CSEPs:
-
Candidate secretory effector proteins
- CMB:
-
Complete media broth
- SBS:
-
Sequencing by synthesis
- RTA:
-
Run time analysis
- CDS:
-
Coding sequences
- TGICL:
-
Transcript gene indices clustering tool
- KAAS:
-
KEGG automatic annotation server
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene ontology
- GH:
-
Glycoside hydrolyase
- CYP:
-
Cytochrome P450
- NGS:
-
Next generation sequencing
References
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D238
Chen S, Su L, Chen J, Wu J (2013) Cutinase: characteristics, preparation, and application. Biotechnol Adv 31:1754–1767. https://doi.org/10.1016/j.biotechadv.2013.09.005
Choi J, Kim KT, Jeon J, Lee YH (2013) Fungal plant cell wall-degrading enzyme database: a platform for comparative and evolutionary genomics in fungi and Oomycetes. BMC Genomics 14(5):S7
Deising H, Nicholson RL, Haug M, Howard RJ, Mendgen K (1992) Adhesion pad formation and the involvement of cutinase and esterases in the attachment of uredo spores to the host cuticle. Plant Cell 4:1101–1111. https://doi.org/10.1105/tpc.4.9.1101
Gueddari EL, Rauchhaus NE, Moerschbacher BM, Deising HB (2002) Developmentally regulated conversion of surface-exposed chitin to chitosan in cell walls of plant pathogenic fungi. New Phytol 156:103–112
Gan PK, Ikeda H, Irieda M, Narusaka ORJ, Narusaka Y, Shirasu K (2013) Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol 197:1236–1249
Jones JD, Dangl JL (2006) The plant immune system. Nature 444:323–329. https://doi.org/10.1038/nature05286
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res 40:D109–D114
Koschorreck K, Liu D, Kazenwadel C, Schmid RD, Hauer B (2010) Heterologous expression, characterization and site-directed mutagenesis of cutinase CUTAB1 from Alternaria brassicicola. Appl Microbiol Biotechnol 87:991–997
Laine MJ, Haapalainen M, Wahlroos T, Kankare K, Nissinen R, Kassuwi S, Metzler MC (2000) The cellulase encoded by the native plasmid of Clavibacter michiganensis ssp sepedonicus plays a role in virulence and contains an expansin-like domain. Physiol Mol Plant 57:221–233
Lara-Márquez A, Zavala-Páramo MG, López-Romero E, Calderón-Cortés N, López-Gómez R, Conejo-Saucedo U, Cano-Camacho H (2011) Cloning and characterization of a pectin lyase gene from Colletotrichum lindemuthianum and comparative phylogenetic/structural analyses with genes from phytopathogenic and saprophytic/opportunistic microorganisms. BMC Microbiol 11:260
Lu L, Rong W, Massart S, Zhang Z (2018) Genome-wide identification and expression analysis of cutinase gene family in Rhizoctonia cerealis and functional study of an active cutinase RcCUT1 in the fungal–wheat interaction. Front Microbiol 9:1813
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35(SUPPL.2):182–185.
Nikolaivits E, Kanelli M, Dimarogona M, Topakas E (2018) A middle-aged enzyme still in its prime: recent advances in the field of cutinases. Catalysts 8:612
O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Vaillancourt LJ (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nature Gen 44:1060–1055
Park J, Lee S, Choi J, Ahn K, Park B, Kang S, Lee YH (2008) Fungal cytochrome P450 database. BMC Genomics 9:402
Prasanth CN, Viswanathan R, Malathi P, Sundar AR (2019) Comparative transcriptome analysis of candidate secretory effector proteins from Colletotrichum falcatum infecting sugarcane. Agri Gene 13:100089
Prasanth CN, Viswanathan R, Krishna N, Malathi P, Sundar AR (2017) Unravelling the genetic complexities in gene set of sugarcane red rot pathogen Colletotrichum falcatum through transcriptomic approach. Sugar Tech 19:604–615
Prasanth CN, Viswanathan R, Malathi P, Ramesh Sundar A. (2021) Development and characterization of genomic SSR marker for virulent strain specific Colletotrichum falcatum infecting sugarcane. 3 Biotech 11: Article number:20. https://doi.org/10.1007/s13205-020-02572-z
Prathima PT, Raveendran M, Kumar KK, Rahul PR, Ganesh Kumar V, Viswanathan R, Ramesh Sundar A, Malathi P, Sudhakar D, Balasubramanian P (2013) Differential regulation of defense-related gene expression in response to red rot pathogen Colletotrichum falcatum infection in sugarcane. Appl Biochem Biotechnol 171:488–503
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clement J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn R (2012) The Pfam protein families database. Nucleic Acids Res 40:D290–D301
Quevillon E, Silventoinen S, Pillai N, Harte N, Mulder R, Apweiler Lopez R (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:6W116–W120
Rahul PR, Ganesh Kumar V, Viswanathan R, Sundar AR, Malathi P, Prasanth CN, Prathima PT (2016) Defense transcriptome analysis of sugarcane and Colletotrichum falcatum interaction using host suspension cells and pathogen elicitor. Sugar Tech 18:16–28
Rao S, Nandineni MR (2017) Genome sequencing and comparative genomics reveal a repertoire of putative pathogenicity genes in chilli anthracnose fungus Colletotrichum truncatum. PLoS One 2017 2(8):e0183567
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–D350
Sathyabhama M, Viswanathan R, Malathi P, Sundar AR (2016) Identification of differentially expressed genes in sugarcane during pathogenesis of Colletotrichum falcatum by suppression subtractive hybridization (SSH). Sugar Tech 18:176–183
Sathyabhama M, Viswanathan R, Nandakumar M, Malathi P, Sundar AR (2015) Understanding sugarcane defence responses during the initial phase of Colletotrichum falcatum pathogenesis by suppression subtractive hybridization (SSH). Physiol Mol Plant Pathol 91:131–140
Singh A, Lal M, Singh MP, Lal K, Singh SB (2000) Variation for red rot resistance in somaclones of sugarcane. Sugar Tech 2(1&2):56–58
Stahl DJ, Schafer W (1992) Cutinase is not required for fungal pathogenicity on pea. Plant Cell 4:621–629. https://doi.org/10.1105/tpc.4.6.621
Viswanathan R (2010) Plant Disease: Red Rot of Sugarcane. Anmol Publishers, New Delhi, India, p 306
Viswanathan R (2021a) Red rot of sugarcane (Colletotrichum falcatum Went). CAB Rev 16: No. 023,. https://doi.org/10.1079/PAVSNNR202116023
Viswanathan R (2021b) Sustainable sugarcane cultivation in India through threats of red rot by varietal management. Sugar Tech 23:239–253. https://doi.org/10.1007/s12355-020-00882-3
Viswanathan R, Padmanaban P, Selvakumar R (2020) Emergence of new pathogenic variants in Colletotrichum falcatum, stalk infecting Ascomycete in sugarcane: Role of host varieties. Sugar Tech 22:473–484
Viswanathan R, Rao GP (2011) Disease scenario and management of major sugarcane diseases in India. Sugar Tech 13:336–353
Viswanathan R, Samiyappan R (1999) Red rot disease in sugarcane: a major constraint for the Indian sugar industry. Sugar Cane 17:9–15
Viswanathan R, Malathi P, Sundar AR, Aarthi S, Prem Kumari SM, Padmanaban P (2005) Differential induction of chitinases and thaumatin like proteins in sugarcane in response to infection by Colletotrichum falcatum causing red rot disease. J Plant Dis Protect 112:417–425
Viswanathan R, Malathi P, Sundar AR, Kaverinathan K, Chhabra ML, Parameswari B, Jothi R (2017) Diversity of Colletotrichum falcatum population in India: comparative virulence at two different agro-climatic regions. Intern Sugar J 119:966–977
Viswanathan R, Prasanth CN, Malathi P, Sundar AR (2016) Draft genome sequence of Colletotrichum falcatum - A prelude on screening of red rot pathogen in sugarcane. J Genomics 4(10):1–3. https://doi.org/10.7150/jgen.13585
Wanjiru WM, Zhensheng K, Buchenauer H (2002) Importance of cell wall degrading enzymes produced by Fusarium graminearum during infection of wheat heads. Eur J Plant Pathol 108(8):803–810
Xiao C, Anderson C (2013) Roles of pectin in biomass yield and processing for biofuels. Front. Plant Sci 4(67)
Yakoby N, Beno-Moualem D, Keen NT, Dinoor A, Pines O, Prusky D (2001) Colletotrichum gloeosporioides pelB is an important virulence factor in avocado fruit-fungus interaction. Mol-Plant-Microbe-Inter 14:988–995
Zhang H, Wu Q, Cao S, Zhao T, Chen L, Zhuang P (2014) A novel protein elicitor (SsCut) from Sclerotinia sclerotiorum induces multiple defense responses in plants. Plant Mol Biol 86:495–511. https://doi.org/10.1007/s11103-014-0244-3
Acknowledgements
The authors are grateful to the Director of ICAR-Sugarcane Breeding Institute, Coimbatore for providing facilities and encouragement.
Author information
Authors and Affiliations
Contributions
RV conceptualised, designed, initiated, and supervised the project. CNP prepared materials and performed whole transcriptome data analysis. CNP performed lab experiments and contributed to data analysis and data interpretations. RV, PM and ARS analysed the interpreted whole information retrieved from the transcriptome analysis. CNP and RV wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Prasanth, C.N., Viswanathan, R., Malathi, P. et al. Carbohydrate active enzymes (CAZy) regulate cellulolytic and pectinolytic enzymes in Colletotrichum falcatum causing red rot in sugarcane. 3 Biotech 12, 48 (2022). https://doi.org/10.1007/s13205-022-03113-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-022-03113-6