
Abstract
Citrus yellow mosaic virus (CYMV) is a member of genus Badnavirus of the family Caulimoviridae. It is the causal agent of citrus yellow mosaic disease in citrus and causes reduction in yield. As the virus is vegetative propagated by grafting, development of high-throughput diagnosis methods based on serological techniques is a prerequisite for production of healthy virus-free planting material. The current study describes the development of polyclonal antibodies raised in rabbits against purified recombinant virion-associated protein (rVAP) encoded by ORF-II of CYMV. The specificity of developed antiserum was evaluated in immunosorbent electron microscopy (ISEM), antigen-coated plate-enzyme linked immunosorbent assay (ACP-ELISA) and immunocapture PCR (IC-PCR). The antiserum specifically reacted up to a dilution of 1:2000 in ACP-ELISA for detection of CYMV-infected plants. The antiserum was validated by screening CYMV-infected plants maintained in the glass house through ACP-ELISA. To the best for our knowledge, this is the first report on production of polyclonal antiserum using recombinant virion-associated protein as fusion protein, which could be used for screening CYMV-infected plants by ELISA and IC-PCR. These immunodiagnostic methods can be effectively employed in routine indexing of citrus and in quarantine process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Citrus yellow mosaic virus (CYMV), a member of genus Badnavirus, family Caulimoviridae is a key constraint in citrus (Citrus sinensis) production which results in reduced yield and affects the fruit quality (Ahlawat 2000). CYMV is bacilliform in shape and size ranges from 30 × 120 to 150 nm. It has double-stranded circular DNA genome of size 7.5 kb. Badnaviruses generally encode three open reading frames (ORFs); however, CYMV has six ORFs; function of ORF I, IV, V and VI is unknown (Huang and Hartung 2001; Anthony-Johnson et al. 2012); ORF III encodes a polyprotein which is cleaved post-translationally by the viral aspartic protease to four functional products, viz., movement protein (MP), coat protein (CP), aspartic protease (PR) and a replicase comprising reverse transcriptase (RT) and ribonuclease H (RNase H) (King et al. 2012). ORF II encodes for virion-associated protein (VAP), a unique protein found in all the members of the family Caulimoviridae, which has a key role in causing infection to the host (Herzog et al. 2000). In plant pararetroviruses, tetramerization of VAP has a vital biological role for interaction with other proteins. VAP acts as the “arm” of virus particle and keeps its C-terminus portion anchored into the capsid shell (Leclerc et al. 2001).
CYMV causes yellow mosaic disease in citrus plants. The disease was initially reported in sweet orange plants with characteristic symptoms of stunting, chlorosis and uniformly distributed leaf mosaic from South India (Dakshinamurti and Reddy 1975; Ahlawat et al. 1996a, b). Incidence of CYMV has been reported in mandarin, sweet orange and acid lime cultivars from Central India where the virus has spread with infected planting material (Ghosh et al. 2007). Based on symptom expression on different citrus cultivars and sequence variations in the viral genome, two distinct strains of CYMV have been recorded (Ghosh et al. 2014). The disease spreads through bud wood grafting and, therefore, development of serological-based techniques is a pre-requisite for indexing and production of clean planting material. Conventionally, antisera for CYMV and most of other Badnaviruses are prepared by purified virus particles (Ahlawat et al. 1996b; Ndowora and Lockhart 2000). However, the concentration of badnaviruses in the infected plants is usually low (Dahal et al. 1998a; Thottappilly et al. 1998) and the process of virus purification is complex. Therefore, production of antisera using purified virus particles will always be a great challenge. Hence, to overcome these issues, use of in vitro expressed virus-specific recombinant coat protein or other structural proteins as an antigen is an alternative method for raising polyclonal antibodies against various plant viruses (Gulati-Sakhuja et al. 2009; Tatineni et al. 2013). In Banana streak MY virus (BSMYV), the precise coat protein sequences were identified (Sharma et al. 2014), and polyclonal antiserum was raised for diagnosing BSMYV. Recently, immunodominant epitopes of two Badnavirus species, Banana streak MY virus and Banana streak OL virus (BSOLV), were identified. For the identified epitopes, anti-peptide antibodies were raised against BSMYV and they successfully detected native virions in ELISA (Vo et al. 2016). Coat protein is not defined in ORF III of CYMV and, hence, recombinant coat protein-based strategy for polyclonal antibodies could not be attempted. In this study, an attempt was made for production of polyclonal antibodies using in vitro expressed recombinant virion-associated protein (rVAP) encoded by ORF II of CYMV for its diagnosis, which will have applications in indexing of large number of citrus plants as well as in quarantine process.
Materials and methods
Cloning of virion-associated protein (VAP)
Specific primer pair for amplification of ORF II of CYMV genome encoding VAP as VAPEcoRI 5′ GCGGAATTCATGAACTATCAGGCTACTGAAA 3′ (forward primer with EcoRI site underlined) and VAP SalI 5′ CGCGTCGACTTGTTGCTGTTTCTTTAGCTTTTC 3′ (reverse primer with SalI site underlined) was designed. Using these primers, VAP region was amplified from the full-length fragment of pUC 18–CYMV clone available in the laboratory of Advance Centre of Plant Virology, ICAR-IARI, New Delhi. The amplified product was visualized in 1.0% agarose gel and eluted using Thermo Scientific GeneJet gel extraction kit (Vilnius, Lithuania, Europe). The gel purified product was sub-cloned into pET28a(+) (Novagen, san Diego, CA, USA) and was transformed into Escherichia coli DH5α strain following the standard protocol (Sambrook and Russel 2001). Transformed products were confirmed by restriction digestion of recombinant plasmids and sequenced for checking orientation and frame of inserts. The pET28a(+) cloned VAP construct was transformed into E. coli strain BL21 (DE3) expression cell line (Stratagene, Lajolla, CA, USA) using the standard procedure (Sambrook and Russel 2001).
In vitro expression and purification
The VAP coding region was expressed in vitro as fusion protein with histidine-binding protein (HBP) in E. coli strain BL21 (DE3). The HBP-rVAP protein was induced with 0.1 M isopropyl-β-d-thiogalactopyranoside (IPTG) to E. coli BL21 (DE3) recombinant plasmid grown at 37 °C for 3 h and analyzed in 12% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). The expressed fusion protein was sonicated from 2 l of 3 h induced bacterial culture and found to be in insoluble fraction, which was examined in 12% SDS-PAGE (Sambrook and Russel 2001). The gel was stained with Coomasie brilliant blue (R250). The desired protein band was excised and destained with 10 ml of 1% ammonium bicarbonate and 10 ml of methanol by keeping in shaker for 15–20 min at room temperature. This step was repeated until the gel becomes colourless. The excised band was crushed with 1% SDS in a pestle and mortar at a ratio of 1:2 (w/v) and centrifuged at 10,000 rpm at 4 °C for 10 min. The supernatant was passed through Whatman filter paper (pre-rinsed with sterile distilled water) to remove polyacrylamide gels. The filtrate was incubated on ice for 10 min to precipitate SDS and centrifuged at 10,000 rpm at 4 °C for 5 min. The supernatant containing protein was collected and the step was repeated for 2–3 times to remove SDS. Finally, the protein was dialyzed against Milli-Q water for 12 h at 4 °C (Vijayanandraj et al. 2013).
Production of polyclonal antiserum
The concentrated mixture of purified protein was used for injection after dialysis. 800 µg of purified CYMV-rVAP fusion protein was emulsified with equal volume of Fruend’s incomplete adjuvant and administered intramuscularly into New Zealander albino rabbit weekly for 3 weeks. A booster injection with 1 mg of purified protein was given after first bleed. Two more bleeds were collected weekly after booster injection. The crude antiserum obtained was mixed with 100% glycerol (1:1 v/v) and stored at − 20 °C.
Enzyme linked immunosorbent assay (ELISA) and immunosorbent electron microscopy (ISEM)
To determine the antibody titre of the recombinant purified protein, extract of infected leaves was used in ELISA. Two-fold dilutions of antigen and a dilution series of antiserum were used in antigen-coated plate (ACP)—ELISA (Clark and Bar-joseph 1984). Blocking was done with 5% bovine serum albumin (BSA) for 1.5 h at 37 °C. Goat-anti-rabbit IgG alkaline phosphatase conjugate (Sigma-Aldrich, India) was used as secondary antibody in ACP-ELISA. Specificity of antiserum (1:100) was confirmed in ISEM using protocol of Milne and Luisoni (1977).
Immunocapture polymerase chain reaction (IC-PCR) for detection of CYMV
Different dilutions (1:250, 1:500 and 1:1000) of the purified polyclonal antiserum raised against CYMV-rVAP was coated (25 µl) on 0.2 ml sterile polypropylene PCR tubes (Axygen, Union city, USA) and kept overnight at 4 °C. IC-PCR was carried out directly in the tubes containing trapped virus particles from the sap preparations of infected leaves with RT/RNaseH region primer pair CYMV RT F—5′ ATGCCATTTGGATTAAAGAAT 3′ (forward primer) CYMV RT R—5′ CCATTTACAGACTCCTCC 3′ (reverse primer) following the standardized thermal cycling conditions with an initial denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 50 °C for 30 s and 72 °C for 1 min and a final elongation step of 72 °C for 10 min. RT/RNase H genomic region is taxonomically important conserved region in the genome of badnaviruses which has been used for identification and PCR-based diagnostics (Yang et al. 2003; King et al. 2012) and, hence, this genomic region was employed for IC-PCR in the present study.
Validation of antibodies for CYMV detection in infected samples and Western blot analysis
A total of 23 citrus leaf samples were taken from the glass house maintained CYMV-infected hosts (ICAR-IARI, New Delhi) for validating the produced antiserum in detecting CYMV infection (Table 1). Crude sap of citrus leaves as antigen at dilution 1:4 (w/v) and antiserum at 1:2000 dilution was used in the study. 100 µl of crude plant sap was coated in a 96-well ELISA plate (NUNC™, Denmark) and the standard procedure of ACP ELISA was performed. Final absorbance at 405 nm was measured with ELISA reader (BIO-TEK instruments, Winooski, USA). Crude leaf extracts of CYMV-infected and healthy leaf prepared by grinding them in 1× PBS, purified protein and antiserum at 1:1000 dilution was used in western blot analysis following the standard protocol (Sambrook and Russel 2001).
Results
Cloning, expression of CYMV-rVAP and purification of recombinant protein
An amplicon of ~ 400 bp of ORF II of CYMV was obtained in PCR amplification from the full-length pUC 18–CYMV clone. It was successfully cloned into pET28a(+) expression vector and was named as CYMV-rVAP construct. Sequencing of CYMV-rVAP in E coli indicated that it contained 418 bp sequence fragment and was in frame. After CYMV-rVAP mobilization into E. coli strain BL21 (DE3), it was induced by adding 0.1 M IPTG (un-induced, 1, 2 and 3 h) at 37 °C, showing the expression of 19kDArVAP as a fusion protein (HBP-rVAP) in the bacterial system (Fig. 1). The expressed protein was observed only in insoluble fraction after sonication. Concentration of 0.1 M IPTG after post-incubation 3 h at 37 °C was best for the expression of fusion protein HBp-rVAP. The recombinant HBP-rVAP was purified by gel elution method and 5 mg of purified protein was harvested and stored at − 80 °C.

Polyacrylamide gel electrophoresis (PAGE) showing in vitro expression of recombinant virion-associated protein (rVAP) of Citrus yellow mosaic virus (CYMV) cloned in pET28a(+) vector and transformed into E. coli BL21 DHE3 expression cell line. The rVAP is over-expressed as a fusion with histidine-binding protein and expresses 19 kDa HBP-rVAP. Lane M: pre-stained marker (Genetix); lane 1, un-induced; lanes 2, 3 and 4: 1, 2 and 3 h after induction with 0.1 mM isopropyl β-d-1,5-thiogalactopyranoside (IPTG)
Production and titre evaluation of polyclonal antiserum
The purified HBP-rVAP protein was injected in rabbit for polyclonal antiserum production. Three bleeds were collected and evaluated for their specific reaction with the crude sap of the infected CYMV leaves by ACP-ELISA. Different dilutions of antiserum for all the three bleeds were taken for evaluation. Serological positive reaction for antigen was observed at 1:64 and above dilutions of antiserum in the second and third bleed (Fig. 2). A low degree of absorbance was observed in the crude sap of healthy sample at the dilution of 1:256 and above. Further, to examine the effective titre value of the antisera, two-fold dilutions of antigen (1:4 and 1:8) and a dilution series of antiserum up to 1:256,000 were taken. An absorbance value of 1.14 was observed in the antiserum dilution 1:2000 as compared to the absorbance for healthy sample of 0.06. The absorbance value declined at further dilutions of antigen and antibody (Fig. 3). Good and differentiating absorbance values were obtained up to the dilutions of 1:4000. In ACP-ELISA, crude sap of CYMV-infected citrus leaves showed 3 times more absorbance values than the healthy sample. A comparison of the absorbance values at different dilutions showed that antiserum dilutions of 1:1000 and 1:2000 can be effectively employed for detection of CYMV in citrus plants. Cross-reactivity of the antiserum was also evaluated with banana sample infected with Banana streak MY virus (BSMYV) and Nicotiana benthamiana crude sap (Table 1). Results showed that the polyclonal antiserum did not react with BSMYV and N. benthamiana plant samples.

ACP-ELISA using different fold dilutions of crude antiserum from different bleeds produced against recombinant HBP-VAP. For titre determination, crude sap from CYMV-infected citrus leaves (grown in glasshouse) was used as antigen at 1:4 dilution. Absorbance values were measured 1 h after substrate addition at 405 nm in ELISA reader. The average value of two replications after subtracting the buffer control reading is shown

ACP-ELISA using different fold dilutions of crude antiserum from third bleed produced against recombinant HBP-VAP. Crude sap from CYMV-infected citrus leaves (grown in glasshouse) was used as antigen at 1:4 and 1:8 dilution to evaluate titer. Absorbance values were measured 1 h after substrate addition at 405 nm in ELISA reader. The average value of two replications after subtracting the buffer control reading is shown
Application of polyclonal antiserum in ISEM and detection of CYMV by IC-PCR
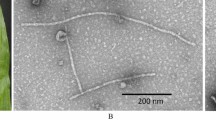
The polyclonal antiserum was used at a dilution of 1:100 for decorating virus particles in ISEM. Decorated bacilliform particles of CYMV were observed in the CYMV-infected citrus plants (Fig. 4a). For IC-PCR-based detection, RT/RNaseH region-specific primers for CYMV genome were used for the analysis. A specific amplicon of size 589 bp was observed when using the HBp-rVAP antiserum at dilutions of 1:250, 1:500 and 1:1000 (Fig. 4b).

Application of HBP-rVAP polyclonal antiserum in immunodiagnostics. a Specific decoration of bacilliform particles of CYMV in immunosorbent electron microscopy (ISEM) using the HBP-rVAP antiserum (1:100 dilution). b Immunocapture PCR (IC-PCR) using RT/RNaseH primers. CYMV-infected citrus samples trapped with HBp-rVAP antiserum. Lane M–O′ generuler ladder mix (Thermo Scientific, USA). Lanes: 1–3 at dilutions of 1:250, 4–6 at dilutions 1: 500, and 7–9 at dilutions 1:1000, respectively. Lanes 1, 4 and 7—amplification of CYMV specific genome of amplicon size ~ 589 bp. Lanes 2, 5, and 8—showed faint amplification of genome size ~ 589 bp. Lanes 3, 6 and 9—showed no amplification in healthy control
Detection of virus in field samples and Western blot analysis
A total of 26 plants were screened for CYMV by ACP-ELISA. The screened samples comprised of 23 citrus samples (collected from Southern and Northern India), 2 banana field samples (infected with BSMYV) and N. benthamiana plant which were used to check the specific reactivity and cross reactivity of the polyclonal antiserum. An absorbance value of three to four-fold was observed in all the CYMV-positive samples when compared to healthy control (Table 1). Specificity of the developed antiserum HBP-rVAP was examined by Western blotting. Crude sap from healthy plant (negative control), infected citrus leaf, and purified protein (positive control) was blotted on the nitro-cellulose membrane. The antiserum HBP-rVAP specifically reacted to the purified protein, showed weak reaction with the CYMV-infected leaf sap and not to the negative control (Fig. 5).

Specific reactivity of developed antiserum HBP-rVAP with the in vitro expressed purified protein used as antigen. Lane 1—crude sap of healthy plant, lane M—Bioeye prestained protein ladder. Lane 2—crude sap of CYMV-infected plant, lane 3—in vitro expressed purified protein
Discussion
Citrus yellow mosaic virus affects several cultivars of citrus like sathugudi (sweet orange), Khasi mandarins, kinnow and pummelo (Dakshinamurti and Reddy 1975; Ahlawat et al. 1985) and causes significant yield loss in citrus production worldwide. The major route of spread of yellow mosaic disease is through infected bud woods, which acts as a vegetative material for propagation of the virus (Lockhart and Olszewski 1993). Therefore, developing diagnostic methods of the graft-transmitted viruses is of great importance, particularly for successful implementation of bud wood certification. For diagnosis of CYMV, some approaches like DNA probes (Korsten et al. 1993), dot-blot hybridization (Borah et al. 2008) and PCR (Baranwal et al. 2005) were available. Although these techniques could detect the virus, there are limitations in terms of their applicability for large-scale indexing, requiring skilled trained manpower and sophisticated laboratory facilities.
On the other hand, ELISA-based techniques are more user friendly and useful for indexing large number of samples. Developing polyclonal antibodies using purified virus preparation as antigen is difficult as it requires large amount of infected tissue and many times the produced antiserum shows non-specific reaction as there are chances of host protein contamination. Badnaviruses usually occur at low concentration which makes it difficult to purify sufficient virus to be used as antigens (Dahal et al. 1998b; Thottappilly et al. 1998). This can be overcome by in vitro expression of viral proteins in bacterial system and development of polyclonal antisera for the effective diagnosis. Recently, Sharma et al. (2014) identified the precise CP gene of Badnavirus in BSMYV—IN1 isolate by comparing the sequences of polyprotein coding region of caulimoviruses. In our studies, expression of CYMV coat protein was unsuccessful. Therefore, an attempt was made to express virion-associated protein for polyclonal antibody production.
ORF II of CYMV yields virion-associated protein (VAP) (Cheng et al. 1996) which is present in all the members of Caulimoviridae (Herzog et al. 2000) and proposed to be a nucleic acid-binding protein (Jacquot et al. 1998). VAP has a tetramer stability which involves in vivo hetromeric interactions with other viral proteins and exhibits some regulatory function (Jacquot et al. 1999). Selvarajan et al. (2016) produced polyclonal antiserum against BSMYV using recombinant VAP as a fusion protein in bacterial system which serologically reacted with the BSMYV-infected banana plants and proved to be antigenic. For the first time, the present study describes successful development of immunodiagnostics against CYMV using recombinant virion-associated protein (rVAP). Tetramerization of VAP has a significant role in all plant pararetroviruses. It acts as the “arm” of virus particle to anchor its C-terminus portion into the virus capsid shell (Leclerc et al. 2001); this property makes the produced recombinant antisera (HBp-rVAP) to be immunogenic and helps in trapping virus particles in serological and nucleo-serological assays. The developed antiserum did not show cross-reactivity with BSMYV confirming the serological diversity among badnaviruses (Lockhart and Olszewski 1993). It is well documented that IC-PCR-based detection is useful to detect the non-integrated viral sequences of all plant pararetroviruses and geminiviruses (Harper et al. 2002). IC-PCR-based detection and Western blot also indicated the specificity of the antisera in reacting only with the CYMV-infected sap and purified protein. Thus, the developed antiserum HBP-rVAP will be useful in ACP-ELISA-based indexing of CYMV in field samples.
To the best of our knowledge, this is the first report on developing immunodiagnostics for detection of CYMV using in vitro expressed recombinant virion-associated protein as a fusion protein in bacterial system.
References
Ahlawat YS (2000) Yellow mosaic. In: Timmer P, Garnsey SM, Graham T (eds) Compendium of citrus diseases, 2nd edn. APS Press, Washington, DC, pp 63–64
Ahlawat YS, Chenulu VV, Viswanath SM, Pandey PK, Bhagabathi KN (1985) Mosaic disease of citrus in India. Curr Sci 54:873–874
Ahlawat YS, Pant RP, Lockhart BE, Srivastava M, Chakraborty NK, Varma A (1996a) Association of a badnavirus with citrus mosaic disease in India. Plant Dis 80:590–592
Ahlawat YS, Varma A, Pant RP, Shukla A, Lockhart BEL (1996b) Partial characterization of a badnavirus associated with citrus yellow mosaic disease in India. In: 13th conference of the international organization of citrus virologists. University of California Press, California, pp 208–217
Anthony-Johnson AM, Borah BK, Sai-Gopal DVR, Dasgupta I (2012) Analysis of full-length sequences of two Citrus yellow mosaic badnavirus isolates infecting Citrus jambhiri (Rough Lemon) and Citrus sinensis L. Osbeck (Sweet Orange) from a nursery in India. Virus Genes 45:600–605. https://doi.org/10.1007/s11262-012-0808-8
Baranwal VK, Singh J, Ahlawat YS, Gopal K, Charaya MU (2005) Citrus yellow mosaic virus is associated with mosaic disease in Rangpur lime rootstock of citrus. Curr Sci 89:1596–1599
Borah BK, Johnson AMA, Gopal DVRS, Dasgupta I (2008) A comparison of four DNA extraction methods for the detection of Citrus yellow mosaic Badnavirus from two species of citrus using PCR and dot-blot hybridization. J Virol Methods 151:321–324
Cheng C, Lockhart BEL, Olszewski NE (1996) The ORF I and II proteins of Commelina yellow mottle virusare virion-associated. Virology 223:263–271
Clark MF, Bar-Joseph M (1984) Enzyme immunosorbent assays in plant virology. In: Maramorosch K, Koprowski H (eds) Methods in virology, vol 3. Academic Press. New York, NY, pp 51–85
Dahal G, Hughes JA, Lockhart BEL (1998a) Status of banana streak disease in Africa: problems and future research needs. Integr Pest Manag Rev 3:85–97
Dahal G, Pasberg-Gauhl C, Gauhl F, Thottappilly G, Hughes JA (1998b) Studies on a Nigerian isolate of banana streak badnavirus: II. Effect of intraplant variation on virus accumulation and reliability of diagnosis by ELISA. Ann Appl Biol 132:263–275
Dakshinamurti V, Reddy GS (1975) Mosaic—a transmissible disorder of sweet oranges. Indian Phytopathol 28:398–399
Ghosh DK, Aglave B, Bhanare K, Baranwal VK (2007) PCR based detection of Citrus yellow mosaic disease from Vidarbha region of Maharastra. Indian Phytopathol 60:520–526
Ghosh DK, Bhose S, Mukherjee K, Aglave B, Warghane AJ, Motghare M, Baranwal VK, Dhar AK (2014) Molecular characterization of Citrus yellow mosaic badnavirus (CMBV) isolates revealed the presence of two distinct strains infecting citrus in India. Phytoparasitica 42:681–689
Gulati-Sakhuja A, Sears JL, Nunez A, Liu HY (2009) Production of polyclonal antibodies against Pelargonium zonate spot virus coat protein expressed in Escherichia coli and application for immunodiagnosis. J Virol Methods 160:29–37
Harper G, Hart D, Moult S, Hull R (2002) Detection of Banana streak virus in field samples of banana from Uganda. Ann Appl Biol 141:247–257
Herzog E, Guerra-Peraza O, Hohn T (2000) The rice tungro bacilliform virus gene II product interacts with the coat protein domain of the viral gene III polyprotein. J Virol 74:2073–2083
Huang Q, Hartung JS (2001) Cloning and sequence analysis of an infectious clone of Citrus yellow mosaic virus that can infect sweet orange via Agrobacterium-mediated inoculation. J Gen Virol 82:2549–2558
Jacquot E, Geldreich A, Keller M, Yot P (1998) Mapping regions of the cauliflower mosaic virus ORF III product required for infectivity. Virology 242:395–402
Jacquot E, Hagen LS, Michler P, Rohfritsch O, Stussi-Garaud C, Keller M, Jacque-mond M, Yot P (1999) In situ localization of cacao swollen shoot virus in agroinoculated Theobroma cacao. Arch Virol 144:259–271
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2012) Virus taxonomy: classification and nomenclature of viruses. In: Ninth report of the international committee on taxonomy of viruses. Elsevier Academic Press, San Diego, pp 385–388
Korsten LG, Sanders HJSu, Garnier M, Bove JM, Kotze JM (1993) Detection of citrus greening- infected citrus in South Africa using a DNA probe and monoclonal antibodies. In: Proceedings of 12th conference IOCV. IOCV, Riverside, pp 224–234
Leclerc D, Stavolone L, Meyer E, Guerra-Peraza O, Herzog E, Hohn T (2001) The product of ORF III in cauliflower mosaic virus interacts with the viral coat protein through its C-terminal proline rich domain. Virus Genes 22:159–165
Lockhart BEL, Olszewski NE (1993) Serological and genomic heterogeneity of banana streak badnavirus: implications for virus detection in Musa germplasm. In: Ganry J (ed) Breeding banana and plantain for resistance to disease and pests. International network for improvement of banana and plantain, Montpellier, France, pp 105–113
Milne RG, Luisoni E (1977) Rapid immuno electron microscopy of virus preparations. In: Maramorosch K, Koprowski H (eds) Methods in virology, vol 6. Academic Press. New York, USA, pp 265–281
Ndowora TCR, Lockhart BEL (2000) Development of a serological assay for detecting serologically diverse banana streak virus isolates. In: Creanen K, Karamura EB, Vulsteke D (eds) Proceedings of the first international conference on banana and plantain for Africa. International Society for Horticultural Science, Leuven, pp 377–388
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual, thirded. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Selvarajan R, Balasubramanian V, Gayathrie T (2016) Highly efficient immunodiagnosis of Episomal Banana streak MY virus using polyclonal antibodies raised against recombinant viral-associated protein. J Phytopathol 164:497–508
Sharma SK, Kumar PV, Baranwal VK (2014) Immunodiagnosis of episomal Banana streak MY virus using polyclonal antibodies to an expressed putative coat protein. J Virol Methods 207:86–94
Tatineni S, Sarath G, Seifers D, French R (2013) Immunodetection of Triticum mosaic virus by DAS- and DAC-ELISA using antibodies produced against coat protein expressed in Escherichia coli: potential for high-throughput diagnostic methods. J Virol Methods 189:196–203
Thottappilly G, Dahal G, Lockhart BEL (1998) Studies on a Nigerian isolate of banana streak badnavirus: I. Purification and enzyme-linked immunosorbent assay. Ann Appl Biol 132:253–261
Vijayanandraj S, Yogita M, Das A, Ghosh A, Mandal B (2013) Highly efficient immunodiagnosis of large cardamom chirke virus using the polyclonal antiserum against Escherichia coli expressed recombinant coat protein. Virus Dis 24:227–234
Vo J, Campbell P, Mahfuz N, Ramli R, Pagendam D, Barnard R, Geering AJ (2016) Characterization of the banana streak virus capsid protein and mapping of the immunodominant continuous B-cell epitopes to the surface-exposed N terminus. J Gen Virol 97:3446–3457. https://doi.org/10.1099/jgv.0.000643
Yang IC, Hafner GJ, Revill PA, Dale JL, Harding RM (2003) Sequence diversity of South Pacific isolates of Taro bacilliform virus and the development of a PCR based diagnostic test. Arch Virol 148:1957–1968
Acknowledgements
We thank ICAR-Outreach project (Grant no. 02) to bear financial assistance for carrying out experiments, Head, Division of Plant Pathology and Director, ICAR-Indian Agricultural Research Institute, New Delhi for providing necessary lab facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kumar, P.V., Sharma, S.K., Rishi, N. et al. Efficient immunodiagnosis of Citrus yellow mosaic virus using polyclonal antibodies with an expressed recombinant virion-associated protein. 3 Biotech 8, 39 (2018). https://doi.org/10.1007/s13205-017-1063-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-017-1063-4