
Abstract
Bacterial symbionts play a critical role in the physiology, ecology and evolution of a diverse range of insects. Such symbionts with unknown roles in the ecology and evolution of their hosts have been reported from archaeococcoid scale insects of family Coelostomidiidae. We examine in detail the bacterial community associated with the remaining species of this family, and calculate the cophylogenetic relationship between the hosts and their symbionts. The 28S ribosomal RNA (rRNA) and mitochondrial cytochrome oxidase I genes were used to reconstruct the host phylogeny while the 16S rRNA gene was used for the bacterial phylogeny. Three well-supported clades were detected within the phylogeny of the monophyletic family Coelostomidiidae. Besides the known symbionts, a novel Sodalis-like symbiont was detected from three of the species. The primary bacteriome inhabiting B-symbiont (Bacteroidetes; ‘Candidatus Hoataupuhia coelostomidicola’) was widespread across the host family. Cophylogenetic comparison using Jungles-based reconciliation analysis and ParaFit statistical test revealed a strongly congruent phylogeny of this symbiont with the host family, with no host-switches and few losses and duplications. A similar pattern was observed across a relatively unrelated neococcoid family that exhibits a different physiology and symbiont community, besides a related Bacteroidetes symbiont. We reconfirm that the B-symbiont is a primary symbiont, owing to its strongly congruent evolution with the host and its bacteriome-inhabiting nature. Our analysis affirms recent suggestions that the Bacteroidetes-affiliated symbionts may have driven the hyper-diversification of scale insects worldwide.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ecological and evolutionary diversification of many eukaryotic organisms has been influenced by their coevolution with endosymbiotic bacteria. This is especially the case for insects with nutrient-limited diets (e.g. aphids and cockroaches), which harbour bacterial symbionts responsible for supplementing their nutrition. A wide variety of insect—bacterial associations are known, including those involving maternally inherited, bacteriome-inhabiting obligate endosymbionts. Due to their specialised yet restrictive environment, these symbionts are separated from external gene pools and their genomes are shaped by host-influenced evolutionary forces (Wernegreen 2002). This can lead to cophylogeny, which is when the host phylogeny is mirrored by the symbiont phylogeny (Page 2003). One such example is the leafhoppers (Cicadellidae) and their primary symbiont Sulcia muelleri, which show congruence in phylogeny across a large and diverse host family (Takiya et al. 2006). Such congruence in host-symbiont phylogenies implies a single event of infection of the last common ancestor with subsequent coevolution between the host and symbiont (Moran et al. 2005). These bacterial symbionts are distinguished from their free-living relatives by their reduced genome size, AT-biased genome composition, lack of recombination, relatively fast rate of sequence evolution, and other genetic factors reflecting their restricted environment (McCutcheon and Moran 2012).
Scale insect families of the world are roughly divided into two main groups, the archaeococcoids and the neococcoids, based on their ancestral lineage (Gullan and Cook 2007). Archaeococcoids are considered remnants of an ancient radiation, while the monophyletic neococcoids are considered ‘advanced’ scale insects (Gullan and Cook 2007). Although little is known about the symbiotic systems of these groups as a whole, some information is available for a select few families representing each group. Archaeococcoids, including members of the families Coelostomidiidae and Monophlebidae, harbour primary Bacteroidetes symbionts inhabiting a bacteriome-like structure (Matsuura et al. 2009; Dhami et al. 2012). Similarly, a prominent and well-studied family of neococcoids, Diaspididae, also contains Bacteroidetes-affiliated symbionts, although a specialised bacteriome-like structure is yet to be found (Gruwell et al. 2012). The diaspidids have a starkly different nutritional physiology compared to typical archaeococcoids. Unlike coelostomidiids that feed on phloem, the diaspidids feed on cellular tissue, and due to a discontinuous digestive system these scale insects do not produce honeydew, the sugary excrement produced by coelostomidiids (McClure 1990). Furthermore, recent analysis of the genome of the diaspidid primary symbiont, Uzinura diaspidicola (host: Aspidiotus nerii), revealed that it is the sole nutritional symbiont of this scale insect, despite the presence of another symbiont Cardinium hertgii (Sabree et al. 2013). Bacteroidetes-affiliated symbionts have been strongly suggested as the primary symbionts of scale insects, although recent work on their cophylogenetic relationship with some host scale insect families suggests that archaeococcoids and neococcoids have acquired these symbionts differentially (Rosenblueth et al. 2012). Overall, archaeococcoids such as coelostomidiids and monophlebids have revealed a greater diversity of symbionts than neococcoids such as diaspidids (Dhami et al. 2012; Matsuura et al. 2009). It is clear that despite sharing a similar primary symbiont, these two groups display distinct symbiotic assemblages.
Our understanding of the symbiotic systems prevalent in scale insects is still in its infancy. Gruwell and colleagues (Gruwell et al. 2007) pioneered research on understanding the family-wide cophylogenetic patterns of Bacteroidetes-affiliated Uzinura symbionts observed in the neococcoid armoured scale family Diaspididae. This study further extends their approach to the archaeococcoid family Coelostomidiidae, which harbours a closely related Bacteroidetes symbiont, in a nutritionally distinct system, involving completely different co-symbionts.
The scale insect family Coelostomidiidae is composed of nine species endemic to New Zealand and five species recorded from South America (Gullan and Cook 2007). For the latter, only morphological data form the basis of the recent systematic placement within Coelostomidiidae (Foldi 2009). In this study, we focus on the New Zealand species. The phylogeny of this family is based on morphological character states alone, most of which are not unique, resulting in low confidence in the phylogeny (Morales 1990). According to Morales (1990), the genus Coelostomidia (six species: C. deboerae, C. jenniferae, C. montana, C. wairoensis, C. pilosa and C. zealandica) is monophyletic and the genus Ultracoelostoma (three species: U. assimile, U. brittini and U. dracophylli) is paraphyletic (Morales 1990). This phylogeny does not explain the observed feeding strategies and behaviour of the species involved. For example, three of the Coelostomidia species, C. deboerae, C. jenniferae and C. pilosa, are notably polyphagous (Morales 1991), feeding on the phloem of a wide range of host plants. In contrast, C. montana, C. wairoensis and C. zealandica, similar to the Ultracoelostoma species, exhibit oligophagy (Gardner-Gee and Beggs 2009). To understand the evolutionary significance of these feeding strategies and their influence on bacterial community composition, it is a logical first step to revisit the phylogenetic relationships of the family Coelostomidiidae.
Given their phloem-feeding habit, it is not surprising that bacteriome-inhabiting B-symbionts (Bacteroidetes) have been detected from a coelostomidiid species, C. wairoensis, in addition to Erwinia-like E-symbionts and Wolbachia (Dhami et al. 2012). The B-symbiont is closely related to the primary symbiont of diaspidids, Uzinura diaspidicola, and to other nutritionally important primary symbionts such as Sulcia muelleri associated with sharpshooters (Cicadellidae) (Dhami et al. 2012). To understand the role of these symbionts, it is critical to ascertain whether any of them are primary, especially the putative primary symbiont: the B-symbiont. The remaining species of the family Coelostomidiidae have not yet been studied.
In the present study, we sequenced a region of the bacterial 16S rRNA gene and a region of the 28S rRNA and cytochrome oxidase I (COI) genes of all nine species belonging to family Coelostomidiidae. The 16S rRNA genes were sequenced after cloning and a snapshot of the bacterial community obtained from each individual species. The cophylogeny analysis was expanded to include published data from the neococcoid family Diaspididae and its associated symbionts (Gruwell et al. 2007). A comparison of patterns of host—symbiont cophylogeny across these two scale insect families was thus possible. Additional gene-based characters, such as AT-composition and relative rates of evolution for the symbionts, were compared to those of their free-living relatives. Our aim was to analyse both the host and symbiont genes in order to (i) identify phylogenetic relationships among New Zealand scale insect species belonging to the family Coelostomidiidae, (ii) examine the bacterial symbiont diversity for each species of the family Coelostomidiidae, and (iii) test for phylogenetic congruence between scale insects of family Coelostomidiidae and their symbiotic bacteria.
2 Methods
2.1 Scale insect collection and identification
Scale insects were collected from 14 sites on the North and South Island of New Zealand, representing multiple samples (from different trees) to multiple populations (from different sites) for each species. The sole exception was Ultracoelostoma assimile, which was found only on a single plant at a single site at Takaka Hill, Nelson (Table 1). Individual scale insects (second instars) were removed from their external test on the host tree using aseptic technique. Ten to 20 individual insects were pooled in a tube containing absolute ethanol to represent a single sample. At each of the sites multiple replicates of such pooled samples were collected. Two to three individuals from each sample were identified using permanent slide mount preparations as per Morales (1991). Voucher slides were accessioned into the New Zealand Arthropod Collection, Landcare Research (Auckland, NZ). Some specimens of C. wairoensis from site Huia were sectioned for transmission electron microscopy (TEM) and fluorescence in situ hybridisation and their symbionts photographed as described in detail in Dhami et al. (2012). Bacteriomes were extracted from Ultracoelostoma brittini from Mt Richardson site and also prepared for TEM and photographed as done in Dhami et al. (2012).
2.2 DNA extraction and PCR amplification
Genomic DNA was extracted from two or more samples per species for each of the nine species, with the exception of U. assimile, as only a single sample was available (Table 1). From each sample, a sub-sample consisting of 3–10 insects was processed using the method of Taylor et al. (2004). Briefly, insect bodies and bacterial cells were disrupted by bead-beating in an ammonium acetate buffer containing chloroform: isoamyl alcohol (24:1). The DNA was precipitated with a 3 M sodium acetate/isopropanol mixture and washed twice in 70 % ethanol, dried and re-dissolved in double-distilled water.
To amplify the 16S rRNA gene, 2 μl of DNA extract were used as template for a PCR reaction using the Bacteria-specific 16S rRNA gene primers 616 V (Juretschko et al. 1998) and 1492R (Kane et al. 1993), which amplify essentially the entire 16S rRNA gene (approx. 1500 bp). Cycling parameters were: an initial denaturing step (94 °C, 6 min); 30 cycles of denaturing (94 °C, 45 s), annealing (55 °C, 45 s) and extension (72 °C, 1.5 min); with a final extension step (72 °C, 10 min).
Genomic DNA from multiple samples per site for each species was used to amplify host DNA for both the 28S rRNA and COI genes. To amplify the 28S rRNA gene, one microlitre of DNA extract was used as template for a PCR reaction using the 28b-S3660 primer pair (Dowton and Austin 1998; Whiting et al. 1997), which amplifies approximately 900 bp of the D2–D3 expansion region. Cycling parameters were: an initial denaturing step (94 °C, 6 min); 30 cycles of denaturing (94 °C, 45 s), annealing (54 °C, 45 s) and extension (72 °C, 1 min); with a final extension step (72 °C, 10 min).
For PCR amplification of the barcoding region of cytochrome c oxidase subunit I gene (COI, approximately 650 bp), the scale insect-specific PcoF1 (5′-CCTTCAACTAATCATAAAAATATYAG-3′) (Park et al. 2010) and the standard insect reverse primer LepR1 (5′-TAAACTTCTGGATGTCCAAAAAATCA-3′) were used. One microlitre of the DNA template was used in the PCR with cycling parameters as follows: initial denaturing step (94 °C, 2 min); 30 cycles of denaturing (94 °C, 40 s), annealing (51 °C, 40 s) and extension (72 °C, 1 min); with a final extension step (72 °C, 5 min).
2.3 16S rRNA gene library construction and sequencing
The amplified 16S rRNA genes from the insect samples were cloned using the pGEM-T Easy vector cloning kit (Promega, USA) following the manufacturer’s instructions. At least two clone libraries per scale insect species were created (except for U. assimile). Samples from a range of geographically distant sites were used for C. wairoensis and C. pilosa for creating up to five clone libraries for each host species. Clone libraries of the amplified 16S rRNA genes were created in an overnight ligation reaction (T4 DNA Ligase at 4 °C) with a pGEM-T Easy vector, followed by transformation of high efficiency Escherichia coli cells (Invitrogen, USA) with the ligated vector. A minimum of 96 successfully transformed E. coli clones per clone library were screened with the vector-specific primers M13F and M13R, and clones containing the correct-sized insert were analysed using amplified ribosomal DNA restriction analysis (ARDRA) (Smit et al. 1997). Restriction enzymes Rsa I and/or Hae III were used in parallel reactions to analyse the diversity of bacterial 16S rRNA genes in each clone library. Digests were performed using 8.8 μL of PCR template with 1 unit of the respective enzyme and 1 μL of reaction buffer for 3 h at 37 °C, followed by 20 min at 65 °C to halt the reaction. Digestion products were visualised on a 3 % agarose gel to obtain ARDRA profiles. Two to three clones representing each ARDRA banding pattern were selected for sequencing, which was performed on an ABI 3130XL capillary sequencer using the vector-specific primers M13F and M13R.
Each of the amplified scale insect genes (28S rRNA gene and COI gene) was also sequenced on an ABI 3130XL capillary sequencer using their respective amplification primers.
2.4 Phylogenetic analysis
All sequences were proof-read and edited using the package Geneious version 5.6.1 (Drummond et al. 2011).
The 16S rRNA gene sequences were checked for chimeras using Bellerophon (Huber et al. 2004), and those that were identified as chimeras were excluded from further analysis. Preliminary phylogenetic relationships were established by submitting these sequences to BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequences of closely related symbionts were downloaded from the NCBI database. The 16S rRNA gene sequences were aligned using the SINA Webaligner (Pruesse et al. 2007) that uses the secondary structure of the 16S rRNA molecule to align sequences. Aligned sequences were manually corrected for misaligned sequences or regions.
The 28S rRNA gene and COI gene sequences were aligned using the MAFFT aligner (G-INS-I, scoring matrix = 200PAM/k = 2) (Katoh et al. 2002). Sequence alignments were manually corrected for misaligned bases.
All sequence alignments were analysed in jModeltest (Posada 2009) to obtain the appropriate model for each of the phylogenies. Eighty-eight models were tested under the BioNJ and fixed ML framework and the Akaike Information Criterion (AIC) was used to rank and select the most appropriate model. The phylogenies were built using PAUP* (Swofford 2003) for likelihood-based analysis and BEAST (Drummond and Rambaut 2007) or *BEAST (Heled and Drummond 2010) for Bayesian inference, using the models selected by jModeltest (Posada 2009). Heuristic searches were performed in PAUP* under the likelihood criterion based on the model suggested by jModeltest. Starting branch lengths were obtained using the Rogers-Swofford approximation method and branch lengths were optimised using the one-dimensional Newton–Raphson algorithm. Starting trees were obtained via stepwise addition with a single tree held at each step. Tree-bisection-reconnection (TBR) branch-swapping algorithm was employed and very short branch lengths (≤1e−008) were collapsed. The analyses were first run with stepwise addition sequence with ten random addition replicates, and then repeated with “as-is” addition sequence, to explore as much tree-space as possible. For the Bayesian inference phylogeny, BEAST analyses with 106 MCMC generations with appropriate burn-in lengths were run 6–8 times. The first two sets of analyses were run with variations of methodology, using Yule speciation process versus Birth-death process of speciation and strict clock versus relaxed clock method with appropriate tree priors. No differences in tree topology were observed and subsequent analyses were set to Yule speciation process in conjunction with strict clock method. Appropriate default tree priors were used in each run. *BEAST was used for construction of the scale insect species tree. This method calculates the multiple species coalescence likelihood of the gene trees embedded in the species tree (Heled and Drummond 2010). Multiple sequences from each scale insect species for each of the two loci analysed, were used to calculate individual gene trees that were used to create the species tree. Independent substitution models were applied to the individual genes based on those suggested by jModelTest. The Yule Model in conjunction with the strict clock method (uniform rates across branches) was applied for species tree construction with species tree prior value = 0.2 (lower = 0.0 and upper = ∞) and shape parameter γ = 0.5. The analysis was run eight times with MCMC chain length = 108 or 5 × 107 and appropriate burn-in. FigTree (Rambaut 2007) was used to visualize and compare trees.
2.5 Cophylogeny analysis
The phylogenetic congruence of the host and symbionts was measured using tree-based, distance-based and likelihood-based methods.
2.5.1 Tree-based reconciliation analysis
TreeMap v3 software (Charleston and Page 2002) was used to perform reconciliation analysis. It depends on a single user-provided tree topology instead of a Bayesian set of trees, thereby not accounting for phylogenetic error. TreeMap uses the Jungles method (Charleston 1998) for mapping a symbiont phylogeny onto a predefined host phylogeny. It is based on the principle of phylogeny reconciliation, which is the process of estimating the historical associations between two phylogenies, mapping the phylogenies and reconciling their differences through evolutionary events (Page and Charleston 1997). Jungles analysis as implemented in TreeMap uses four types of events: cospeciation (when host and symbiont lineages speciate concurrently), duplication (the symbiont lineage speciates independently of the host, but both new symbionts remain on the host), lineage sorting/loss (the symbiont lineage does not speciate when its host lineage does, or becomes extinct) and host switching (the symbiont lineage speciates, but does not follow host speciation by switching to another host). This method finds the solutions by cost-minimization of mapping the dependent symbiont phylogeny, P, on the independent host phylogeny, H (Charleston and Page 2002). In case of an incongruent phylogeny, the program uses host switching as the underlying mechanism, as displayed in the case of a facultative parasite. Optimal solutions explaining the symbiont phylogeny, given the host phylogeny, are calculated through a heuristic search. It is computationally prohibitive to find all possible solutions (Ovadia et al. 2011), therefore the Jungles analysis was run for 25 generations to calculate the optimal solution, as prescribed by the program manual. Identical solutions were obtained by repeating the analysis multiple times. The distant outgroups used for tree construction were removed to restrict the reconciliation analysis to the scale insect families of interest (Coelostomidiidae, Monophlebidae and Diaspididae). The statistical significance of overall congruence of host and symbiont phylogeny using repeated independent randomization tests (Monte Carlo, n = 100) was also computed (Charleston and Page 2002). The resulting p-values denote the expected proportion of times the randomised symbiont phylogeny can be mapped onto the original host phylogeny as well as the original symbiont phylogeny. In addition, a Patristic Distance Correlation Test was also run for obtaining p-values for individual nodes on the symbiont and host phylogenies.
2.5.2 Tree distance-based analysis
ParaFit was used to test the hypothesis of coevolution between the 28S rRNA (host) —16S rRNA (symbiont) datasets and the COI (host) —16S rRNA (symbiont) datasets. The null hypothesis (H0) of the global test is that the evolution of the host and symbiont, as described by their phylogenetic trees and host-symbiont associations, has been independent (Legendre et al. 2002). The global test involves random permutations of the host associated with each symbiont, since symbionts are present in the host and not the opposite (Legendre et al. 2002). In the absence of a coevolutionary association, each symbiont should be able to infect hosts selected at random. This forms the null hypothesis of the global test. To test the individual host-symbiont association links, two statistics were used (ParafitLink1 and ParafitLink2), based on the idea that the global statistic should decrease in value if we remove an association link that represents an important contribution to the host-symbiont relationship (Legendre et al. 2002). The maximum possible value would occur when the host and symbiont phylogenies are fully congruent. The genetic distance matrices for each of the datasets employed in this program were obtained using PAUP* under the parsimony criterion. The distance matrices were formatted and reordered by taxa in the program Patristic v1.0 (Fourment and Gibbs 2006). The reordered distance matrices were entered into the program DistPCoA (Legendre and Anderson 1998) to calculate principal coordinates. The principle coordinate matrices, along with the host-symbiont association matrix, were entered into ParaFit to evaluate the null hypothesis of independent evolution of host and symbiont. Probabilities were calculated after 999 permutations for each of the ParafitLink statistic and the Global Test.
2.5.3 Likelihood-based analysis
The Shimodaira-Hasegawa likelihood-based test (SH test) evaluates whether some trees are better than others at explaining the sequence data (Shimodaira and Hasegawa 1999). We tested each dataset against both host and symbiont trees using 10,000 replicates in the RELL (re-estimation of log likelihoods) approximation (one-tailed test) using the best-fit AIC model in PAUP*.
2.6 Characterisation of the symbiont 16S rRNA genes
The AT composition of the symbiont 16S rRNA gene sequences was calculated using the Statistics tool implemented in Geneious (Drummond et al. 2011). Published sequences for free-living/non-symbiotic relatives of the symbionts were downloaded from the Ribosomal Database Project (RDP), SSU database (Maidak et al. 2001). Relative rates of evolution were calculated for each of the symbiont 16S rRNA genes and compared with that of a free-living/non-symbiotic relative. Tajima’s relative rates test (Tajima 1993) was implemented in software Mega5 (Tamura et al. 2011).
3 Results
3.1 Phylogeny of family Coelostomidiidae
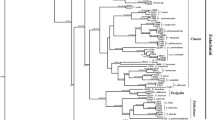
Molecular data from two genetic loci together provide a good resolution of the phylogeny of the New Zealand Coelostomidiidae (Fig. 1). The nine scale insect species can be split into three clades, namely oligophagous Coelostomidia clade 1 (C. montana, C. wairoensis and C. zealandica), polyphagous Coelostomidia clade 2 (C. pilosa, C. jenniferae and C. deboerae) and monophagous Ultracoelostoma clade 3 (U. brittini, U. assimile and U. dracophylli). These three clades were consistent throughout the analyses, independent of the method employed. The cytochrome oxidase I gene tree (Bayesian and likelihood analyses) reconstructs the phylogeny as (Coelostomidia clade 2, (Coelostomidia clade 1, Ultracoelostoma clade 3)) while the 28S rRNA gene tree (Bayesian and Likelihood analysis) predicts the phylogeny as (Coelostomidia clade 1, (Coelostomidia clade 2, Ultracoelostoma clade 3)) (data not shown). The two genes were also concatenated and resulted in the phylogeny as predicted by the 28S rRNA gene, possibly biased due to the use of a longer region of the 28S rRNA gene. We obtained a species tree with the individual gene trees embedded in it through coalescence, using *BEAST (Heled and Drummond 2010). The resulting species tree (Ultracoelostoma clade 3; posterior = 0.9904, (Coelostomidia clade 1, Coelostomidia clade 2; posterior = 0.9023)) is shown in Fig. 1.

Species tree based on 28S rRNA gene and COI gene for family Coelostomidiidae. The associated bacterial symbiont diversity (based on 16S rRNA gene libraries) is represented by the pie charts adjacent to each scale insect species. *Denotes the node where the E-symbiont may have been lost in the scenario that the B-, S- and E-symbiont were all acquired via the last common ancestor. Bayesian posterior probabilities are labelled at the nodes
3.2 Bacterial symbiont community structure
In this study, four different insect-associated bacteria were detected in the coelostomidiid clone libraries. These were the B-symbiont (Bacteroidetes), novel associate: Sodalis-like symbiont (Gammaproteobacteria) (hereafter referred to as the S-symbiont), Erwinia-like symbiont (Gammaproteobacteria) or E-symbiont (Dhami et al. 2012) and Wolbachia sp. (Alphaproteobacteria). In addition, plant-associated or environmental bacteria were found in some of the clone libraries at low numbers (Supplementary Table 1).
The B-symbiont (Fig. 2) was notably the most recurrent symbiont (present in 7/9 species) and when present was also the dominant symbiont in most cases (% distribution of sequences ranging from 46 % to 100 %). An exception to this was in the presence of the S-symbiont, where B-symbiont densities were either reduced (for example 8.38 % in C. jenniferae), or absent altogether (Supplementary Table 1). S-symbiont was the most dominant symbiont type when present (3/9 species), with % distribution of sequences ranging from 50 % to 99 % (Supplementary Table 1). E-symbiont had a relatively narrow % distribution, ranging from 30 % to 37 % when present (3/9 species). Wolbachia was present at medium to low relative densities in almost all Coelostomidia spp. (exception: C. jenniferae). At most, in any of the species, a maximum of three types of bacterial symbionts were found to coexist. A large multi-lobed bacteriome embedded with B-symbionts was localised in the abdomen region of C. wairoensis as observed with fluorescence in situ hybridisation (Fig. 2a and b). Due to the orientation of the insect during sectioning, it is difficult to acquisition both bacteriomes on a single section. From transmission electron microscopy of C. wairoensis, we were able to obtain images of multiple symbiont morphotypes, of which the B-symbionts were localized within the bacteriome (Fig. 2c), as reported previously (Dhami et al. 2012). The additional morphotypes could be E-symbionts or Wolbachia, as both species are reported in C. wairoensis. Due to size restrictions, only the bacteriomes of U. brittini could be used for TEM and were found to be populated with B-symbionts (Fig. 2d), very similar in size and shape to those found previously in C. wairoensis (Dhami et al. 2012).

B-symbiont of coelostomidiid scale insects: a shows fluorescently labelled B-symbiont in a longitudinal section of Coelostomidia wairoensis. A single bacteriome can be observed in this section, although a pair of such bacteriomes is present in the abdominal region of the whole organism. The Bacteroidetes–specific CF319a probe is labelled with Cy5 and the non-EUB negative-probe is labelled with Fluos. b Subtraction of the two filters shows the bright cyan-coloured bacteriome, full of B-symbiont bacterial cells. c Transmission electron micrograph of the bacteriome located in whole insect sections of C. wairoensis shows the morphology of the B-symbiont cells inhabiting the bacteriome. d Transmission electron micrograph of the bacteriome of Ultracoelostoma brittinii shows the morphology of the B-symbiont cells inhabiting the bacteriome
3.3 Insect host and Bacteroidetes cophylogeny
The B-symbiont was selected for detailed cophylogeny analyses, due to its high prevalence across Coelostomidiidae, as well as previously suggested putative primary status (Dhami et al. 2012). Forty-two host-symbiont pairs for the 28S rRNA-16S rRNA dataset and 22 host symbiont pairs for the COI-16S rRNA dataset were analysed. These data spanned a host range including the families Monophlebidae, Diaspididae (armoured scales), Cicadellidae (sharpshooters and cicadas), Coccinellidae (ladybird beetles) and Blattidae (cockroaches), in addition to Coelostomidiidae (Supplementary Table 2). The resulting tanglegram indicated an overall mirroring of the host-symbiont phylogenies across all the major clades (Fig. 3). The p-values of individual nodes derived from the Patristic Distance Correlation Test implemented in TreeMap indicated strong support for congruence between host and symbiont phylogenies at the family level, tribe level and in several subclades (Fig. 3). Statistical significance of overall congruence for both datasets was observed (28S rRNA—16S rRNA: p = 0 and COI—16S rRNA: p = 0). The major clades with significant p-values in the tanglegram also had good posterior probability support for the lineages, as indicated in Fig. 3. Furthermore, the jungles analysis of the 28S rRNA—16S rRNA dataset for the scale insect families (total of 39 host-symbiont pairs from families Coelostomidiidae, Monophlebidae and Diaspididae) predicted a single optimal solution with 52 cospeciation events, 24 duplications, zero host-switches, and 44 losses. Of these, family Coelostomidiidae is responsible for four cospeciation events, a single duplication and two loss events (Fig. 4). Similar results were observed for the COI—16S rRNA dataset, with 36 cospeciation events, 10 duplications, zero host switches and 16 losses. Overall the data suggest that the scale insect families have coevolved with their respective Bacteroidetes symbionts.

Tanglegram of host insect phylogeny (28S rRNA gene) and symbiont phylogeny (16S rRNA gene) depicting the host-symbiont associations. Red dots indicate significance of congruence between host and symbiont phylogenies. Main host families and Bacteroidetes symbionts involved are labelled. Posterior probabilities are indicated at major nodes. A list of sequences and their identities is provided in the Supplementary Table 2

A representative reconciled tree for Bacteroidetes symbionts associated with scale insects (Coelostomidiidae, Diaspidiidae and Monophlebidae). This is simplified from the optimal solution reconstruction via jungles analysis in TreeMap v3. Host phylogeny is denoted by black tree and symbiont phylogeny by grey tree. The host-symbiont links are given at the tips. No host switching events were predicted. Sequence accession numbers are provided in Supplementary Table 2
The Parafit test results were corroborative for the 16S rRNA—28S rRNA and 16S rRNA— COI gene datasets. The global test detected a highly significant (p = 0.001) overall coevolutionary structure. However, the tests on the individual links indicated a mixed situation where some parts of the two trees were congruent whereas other parts were not. In such a situation it is suggested that ParaFitLink1 is the recommended statistic, as the type 1 error on the random links (ParaFitLink 2) is inflated (Legendre et al. 2002). All of the Coelostomidiidae host—B-symbiont links (ParaFitLink 1) using 28S rRNA—16S rRNA were significantly congruent, while more than half of those observed for Diaspididae were not (Table 2). This indicates a relatively stronger congruence in the host-symbiont phylogeny of family Coelostomidiidae.
The SH test indicated that there is no significant difference between the most likely topology supported by the host (28S rRNA gene) and the Bacteroidetes (16S rRNA gene) dataset (P = 0.4712). The SH test is a very conservative test as compared to other likelihood-based topology tests (Buckley 2002). This result further indicates that the scale insects from families Coelostomidiidae, Diaspididae and Monophlebidae have coevolved with their Bacteroidetes symbionts.
3.4 Cophylogeny of Coelostomidiidae and other symbionts
Host phylogeny based on the 28S rRNA and COI genes was compared with the respective symbiont phylogeny based on the 16S rRNA gene. The tanglegram for Wolbachia and coelostomidiid host scale insects revealed a completely incongruent pattern (Fig. 5). None of the nodes were significantly congruent. The jungles analyses revealed that there were six cospeciation events, three duplications, one host switch and six losses. The SH test and Parafit analyses were not applied to this dataset due to the small number of taxa involved.

Schematic tanglegrams for the host (Coelostomidiidae) and symbiont (Wolbachia, S-symbiont and E-symbiont) phylogenies
The tanglegram (Fig. 5) for S-symbiont and coelostomidiid hosts revealed that there was some congruence in the data as the phylogeny of the symbiont relationships was reflective of the host species relationships. The SH test and Parafit analyses were not applied to this dataset as they would not provide meaningful answers due to the small number of taxa involved.
The tanglegram (Fig. 5) for the E-symbiont and coelostomidiid hosts revealed that there was some congruence amongst the two phylogenies, while none of the nodes were found to be of significant congruence. Like the S-symbiont, the absence of this symbiont from two-thirds of the host family reduces the applicability of the SH and Parafit analyses to this dataset.
3.5 Genetic properties of symbiont 16S rRNA gene
The 16S rRNA gene of the B-symbiont showed a significantly slower rate of evolution when compared to that of a free-living/non-symbiotic relative, irrespective of whether the reference outgroup organism was closely or distantly related (Table 3). This analysis was repeated with multiple reference outgroups and free-living/non-symbiotic relatives as the power of relative rates tests depends highly on the selection of outgroup and free-living relatives (Bromham et al. 2000). The E- and S-symbionts did not show dissimilar rates of evolution to their nearest free-living/non-symbiotic relatives (Table 3). In the case of Wolbachia, variable results were observed, with different relatives. Most closely related organisms in the group Rickettsiaceae are either saprophytic or parasitic, therefore making it difficult to select an appropriate free-living/non-symbiotic organism.
The 16S rRNA gene sequences from the B-symbiont exhibited AT contents ranging from 50.4 % to 52.4 % (average = 51.7 %), which is markedly higher than the average 45.65 % AT content of free-living/non-symbiotic Bacteroidetes (data source RDP, n = 498). On the other hand, the 16S rRNA gene sequences of S-symbiont and E-symbiont had an AT content of 44.5 % and 44 % respectively, which was similar to the values in free-living Gammaproteobacteria. Interestingly, AT composition of the 16S rRNA gene sequences of Wolbachia was 51.6 %, which is considerably lower than that of free-living/non-symbiotic Alphaproteobacteria (63 %) (data source RDP, n = 1399).
4 Discussion
4.1 Phylogeny of the New Zealand Coelostomidiidae
This study reports the first DNA-based species tree of the family Coelostomidiidae (New Zealand), based on cytochrome oxidase I barcoding region and D2–D3 expansion region of the 28S rRNA gene. Previous attempts at reconstructing the phylogenetic relationships of this family have either been based on morphological characters alone (Morales 1990) or have not included data from all nine species (Gullan and Cook 2007). We were able to obtain strong support (Bayesian posteriors) for monophyly of the three ecologically distinct groupings in this family. Gene tree discordance from species trees is widespread and is often attributed to incomplete lineage sorting (Degnan and Rosenberg 2009; Pollard et al. 2006). In such an event, a consensus or concatenated tree can be used. Both these approaches have several flaws and can lead to an incorrect species tree, especially if more than three taxa or a low number of samples are involved (Degnan and Rosenberg 2009). Concatenation often fails to derive the correct species tree as it assumes a single rate of evolution for all of the concatenated genes. This being the case, we used *BEAST which co-estimates multiple gene trees embedded in a shared species tree (Heled and Drummond 2010). This method performs better than most other multilocus methods even if low numbers of samples per species are present (Heled and Drummond 2010). The phylogeny obtained in this study agrees with the observed ecological and evolutionary characters of the coelostomidiids and differs significantly from the morphology-based phylogeny proposed by Morales (1990).
4.2 Bacterial symbiont community structure associated with Coelostomidiidae
The bacterial communities associated with coelostomidiid scale insects in New Zealand are diverse in more than one way—phylogenetically divergent from each other, in addition to being affiliated with functionally dissimilar groups of bacteria. This study reports four major bacterial symbiont types, of which three were previously reported from C. wairoensis (Dhami et al. 2012), and several minor bacterial associates. The B-symbiont (Bacteroidetes, affiliated with Sulcia, Blattabacterium and Uzinura) was previously postulated to be a primary symbiont and was generally present with high infection densities in this study. Its absence from two of the species in this family and low infection density in a third species was correlated with the presence of another insect-specific symbiont, the Sodalis-like or S-symbiont. Sodalis sp. is a beneficial secondary symbiont of a number of insects (Wren 2002; Kaiwa et al. 2010; Dale and Maudlin 1999). Sodalis sp. and Sodalis affiliates replaced ancient primary symbionts in Dryophthoridae weevils (Conord et al. 2008) and may be a source for symbiont replacements in adelgids (Hemiptera: Adelgidae) (Toenshoff et al. 2012). Their presence in some of the species of Coelostomidiidae is suggestive of symbiont replacement. Based on the superficial pattern of coevolution observed, in addition to similar AT-composition and relative rates of evolution with free-living bacteria (based on the 16S rRNA gene), it is likely that symbiont replacements are relatively recent and limited to a few species. Absence of the B-symbiont from the two other species was checked using additional clone libraries where possible. However, due to a large geographical spread of some of the species and unknown dispersal mechanism, it is difficult to rule out the possibility of localised differences in symbiont diversity. Ultracoelostoma assimile, which is virtually indistinguishable from U. brittini based on COI gene sequence (Ball and Armstrong 2007), morphological characters and habitat (Morales 1991), did not harbour the B-symbiont. This species is smaller in size (U. assimile: 2.0–3.8 mm in length × 1.8–3.1 mm wide; U. brittini: 4.4–6.0 mm in length × 3.6–5.2 mm wide) and appears to be rare compared with U. brittini, although more work is needed to resolve its distribution. We hypothesise that the reduced size and apparent low biotic success of U. assimile is correlated with its loss of B-symbiont.
Wolbachia, which is a well-known arthropod reproductive parasite, was also widespread across this family. Its variability amongst different populations of the same species, lack of congruent phylogeny or symbiont-like 16S rRNA gene characters, suggest that it is not a mutualistic symbiont (Fig. 5, Supplementary Table 1). As a reproductive parasite, it is capable of infecting individual populations horizontally, thereby obliterating any clear host-symbiont relationship. Once acquired into a population, it is known to be capable of vertical transmission through generations (Stouthamer et al. 1999).
The presence of the Erwinia-like gammaproteobacterium (Enterobacteriaceae) or E-symbiont was confirmed from another two species in family Coelostomidiidae. The genus Erwinia is little known as an insect symbiont and is better known as a plant pathogen (Alfano and Collmer 1996). Enterobacteriaceae–allied bacteria (of unknown function) have been reported from a range of archaeococcoid and neococcoid scale insect families including Monophlebidae, Ortheziidae, Diaspididae, Putoidae, Pseudococcidae, Coccidae and Lecanodiaspididae (Matsuura et al. 2009; Rosenblueth et al. 2012; Gruwell et al. 2010). E-symbionts reported are a sister group to a number of insect symbionts such as Sodalis, Buchnera, and Blochmannia. Little can be inferred about the role of this species in Coelostomidiidae as only superficial coevolutionary patterns, relative rates of evolution and AT content similar to free-living relatives were observed from the available data. Recent analysis of the coevolutionary relationship of enterobacterial symbionts with their respective scale insect host families (Ortheziidae, Monophlebidae, Pseudococcidae, Coccidae and Diaspididae) suggested an incongruent relationship, with many duplication and host switch events predicted in the symbiont phylogeny (Rosenblueth et al. 2012). Although this study only included a few species from each family analysed, it does provide a wider context for the patterns of congruence that we observed in Coelostomidiidae, with respect to their E-symbionts. A thorough Coccoidea-wide survey for E- and S-symbionts is needed to further clarify their role as symbionts of scale insects.
4.3 Coevolution of scale insects and Bacteroidetes symbionts
Symbionts belonging to the phylum Bacteroidetes are widespread amongst insects and feature in some of the most ancient insect-bacterial obligate mutualisms known, e.g. between Sulcia muelleri and sharpshooters (Hemiptera: Cicadellinae), dated to at least 260 million years ago (McCutcheon and Moran 2007). Such symbionts strictly coevolve with their host and display several degenerative genomic characters, such as small genome sizes, AT-biased nucleotide composition, and high relative rates of molecular evolution (see Wernegreen 2002 for review). Members of the phylum Bacteroidetes have been reported from several scale insect families, namely Coelostomidiidae, Monophlebidae, Ortheziidae (Archaeococcoids) and Diaspididae, Pseudococcidae and Eriococcidae (Neococcoids) (Dhami et al. 2012; Matsuura et al. 2009; Gruwell et al. 2005, 2007). This study combines the published literature on the 16S rRNA gene of the Bacteroidetes symbiont from the well-studied neococcoid family Diaspididae and archaeococcoid family Coelostomidiidae (sequences from this study) and available 28S rRNA gene and cytochrome oxidase gene data of the respective hosts to compare the cophylogenetic patterns across these unrelated groups. Not surprisingly, strong congruence with a strong Bayesian support for major clades is observed between host and symbiont phylogenies. The symbiont phylogeny closely follows that of the host in terms of divergence of sister taxa. For example, the Coelostomidiidae are more closely related to Monophlebidae (Coelostomidiidae + Monophlebidae clade posterior = 0.99) than Diaspididae and the corresponding symbiont phylogeny displays this same pattern (B-symbiont + symbiont of Icerya brasiliensis (DQ868792) clade posterior = 0.99) (Fig. 3). Also, within the family Diaspididae the bifurcation of the two subfamilies (Aspidiotinae + Diapidinae clade, posterior = 0.99) is mirrored in the phylogeny of the symbiont (Uzinura diaspidicola clade posterior = 0.99) (Fig. 3). This corroborates previous work on cospeciation of armoured scales (Diaspididae) and Uzinura diaspidicola (Gruwell et al. 2007) and extends the scale insect-Bacteroidetes cophylogeny to include the family Coelostomidiidae. The jungles analysis reconstructs the historic coevolutionary patterns based on the host and symbiont phylogenies and the absence of host-switches within the Coelostomidiidae clade adds to the “primary symbiont” status of the B-symbiont (Fig. 4). The TreeMap analysis relies heavily on a single optimal tree and therefore future analysis using multiple symbiont genes and additional host genes will provide a more resolved and robust pattern. In this study, additional support for the TreeMap analysis was obtained through the use of statistically robust tests such as Parafit and the highly conservative SH-Test. The strong statistical support from the Parafit analysis of global and individual link congruence of B-symbiont within the family Coelostomidiidae further strengthens our conclusion that the B-symbionts have congruent phylogenies with their hosts. It did however reveal that not all the host-symbiont associations in family Diaspididae were significant. Additionally, this analysis provides further support for the recent work on the co-speciation of Bacteroidetes-affiliated symbionts and their scale insect hosts. The well-supported loss event followed by multiple duplications (Fig. 4) that separates the coelostomidiid clade and diaspidid clade further supports the hypothesis that their symbionts may have been acquired independently from the ancestral lineage or suffered a loss and then re-acquisition by the diaspidids (Rosenblueth et al. 2012). This suggests that the scale insects have at least two lineages of closely related Bacteroidetes symbionts, in addition to those of pseudococcids that are quite distinct from either. In both these lineages, strict congruence suggests that the Bacteroidetes symbionts are indeed the primary symbionts of these scale insects.
Limited information on the genetic characteristics of an organism can be derived from its 16S rRNA gene alone. Nevertheless, it has been used repeatedly as an indicator for contrasting AT-composition and to gauge the relative rates of evolution. The AT bias observed for the B-symbiont 16S rRNA gene is consistent with that observed for other symbionts. The significantly slower relative rate of evolution is however unusual for this symbiont. These characters must be thoroughly explored through whole genome sequencing in order to compare the genomic AT-composition and net rates of evolution with those of well-described primary symbionts.
These results, in conjunction with previous work on C. wairoensis, suggest that the B-symbiont is likely to be an obligate primary symbiont, as it not only has a congruent phylogeny with the host family, but it shows the genetic features characteristic of a maternally inherited symbiont. We provide further evidence that these scale insect families have inherited these Bacteroidetes symbionts independently from a common ancestor, subsequently evolving on parallel trajectories to exhibit the strongly congruent symbiont phylogenies. This illustrates the importance of the close association of these symbionts to the diversification of their scale insect hosts.
4.4 ‘Candidatus’ status for the B-symbiont
The B-symbionts are unique to the scale insect family Coelostomidiidae. The next closest described symbiont Uzinura diaspidicola (Accession # DQ868793–DQ868798 vs. each of B-symbiont sequences from all seven coelostomidiid hosts) only shares 86–86.8 % 16S rRNA sequence similarity with this symbiont (based on pair-wise similarity over the entire sequence length using BLAST). As suggested by other studies estimating the origin of Bacteroidetes symbionts in scale insect families and reiterated in this study, the multiple acquisition events preceded by loss of ancestral Bacteroidetes symbiont in the neococcoids (Diaspididae) (Rosenblueth et al. 2012), may explain the unique clade formed by the B-symbionts of Coelostomidiidae. Partially characterized and uncultivated microorganisms may be given the designation ‘Candidatus’, therefore we propose to name the lineage corresponding to the B-symbionts of family Coelostomidiidae as ‘Candidatus Hoataupuhia coelostomidicola’ gen. nov., sp. nov. ined.. Hoataupuhia (Hoa. tau. pu’.hia) Māori n. companion in symbiosis (from: hoa. Māori n. companion, friend; taupuhipuhi n./adj. = symbiosis/symbiotic, used for organisms (especially of different species) living together but not necessarily in a relation beneficial to each); N.L. neut. N. Hoataupuhia; coelostomidicola (coel. o. sto. mi. di. cola. N.L. fem. pl. n. Coelostomidiidae a scale insect family; N.L. gen. pl. n. coelostomidicola of the Coelostomidiidae, referring to the host family of the scale insects it has been identified from).
5 Conclusion
We provide evidence for a complex evolutionary history between multiple bacterial symbionts and coelostomidiid scale insects. Symbiotic associations of both congruent and incongruent nature are present, possibly fulfilling functionally distinct roles. This type of symbiotic system is quite distinct from that of the neococcoid family Diaspididae, however further work is needed to discover functional differences within these two symbiotic systems. There is strong evidence for the strictly congruent relationship between the B-symbiont ‘Candidatus Hoataupuhia coelostomidicola’ and the hosts, but little evidence of congruent phylogeny of the reproductive parasite Wolbachia. The presence of multiple symbionts in conjunction with the primary symbiont or through replacement of the primary symbiont, especially in the polyphagous species, indicates that this advanced character may have evolved due to access to multiple symbiotic capabilities. However, this also raises question of whether this symbiotic system is truly ancestral to that of neococcoids with fewer symbionts. Our results are consistent with bacterial symbionts facilitating niche extension, which results in hyper-diversification of certain groups of insects. This study sets the groundwork for the future exploration of Bacteroidetes as a primary driver of evolution in scale insects.
References
Alfano JR, Collmer A (1996) Bacterial pathogens in plants: life up against the wall. Plant Cell 8(10):1683–1698
Ball S, Armstrong K (2007) Using DNA barcodes to investigate the taxonomy of the New Zealand sooty beech scale insect. DOC research & development series, vol 287. Bio-Protection Research Centre, New Zealand
Bromham L, Penny D, Rambaut A, Hendy MD (2000) The power of relative rates tests depends on the data. J Mol Evol 50(3):296–301
Buckley TR (2002) Model misspecification and probabilistic tests of topology: evidence from empirical data sets. Syst Biol 51(3):509–523. doi:10.1080/10635150290069922
Charleston M (1998) Jungles: a new solution to the host/parasite phylogeny reconciliation problem. Math Biosci 149(2):191–223
Charleston M, Page R (2002) TreeMap. v. 2.0. 2. Software distributed by authors
Conord C, Despres L, Vallier A, Balmand S, Miquel C, Zundel S, Lemperiere G, Heddi A (2008) Long-term evolutionary stability of bacterial endosymbiosis in Curculionoidea: additional evidence of symbiont replacement in the Dryophthoridae family. Mol Biol Evol 25(5):859–868. doi:10.1093/molbev/msn027
Dale C, Maudlin I (1999) Sodalis gen. nov. and Sodalis glossinidius sp. nov., a microaerophilic secondary endosymbiont of the tsetse fly Glossina morsitans morsitans. Int J Syst Bacteriol 49(1):267–275
Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24(6):332–340
Dhami MK, Turner AP, Deines P, Beggs JR, Taylor MW (2012) Ultrastructural and molecular characterization of a bacterial symbiosis in the ecologically important scale insect family Coelostomidiidae. FEMS Microbiol Ecol 81(3):537–546. doi:10.1111/j.1574-6941.2012.01378.x
Dowton M, Austin AD (1998) Phylogenetic relationships among the microgastroid wasps (Hymenoptera: Braconidae): combined analysis of 16S and 28S rDNA genes and morphological data. Mol Phylogen Evol 10(3):354–366
Drummond A, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7(1):214. doi:10.1186/1471-2148-7-214
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2011) Geneious v 5.6. Available from http://www.geneious.com
Foldi I (2009) Archaeococcoid scale insects (Hemiptera: Coccoidea) from the tropical high mountains of the Andean Cordillera, South America. Zootaxa 2300:1–38
Fourment M, Gibbs MJ (2006) PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol Biol 6 (1). doi:10.1186/1471-2148-6-1
Gardner-Gee R, Beggs JR (2009) Distribution and abundance of endemic coelostomidiid scale insects (Hemiptera: Coelostomidiidae) in Auckland forests, New Zealand. N Z J Ecol 33(2):138–146
Gruwell ME, Von Dohlen CD, Patch K, Normark BB (2005) Preliminary PCR survey of bacteria associated with scale insects (Hemiptera: Coccoidea). Proceedings of the Tenth International Symposium on Scale Insect Studies, Adana, Turkey, 19–23 April 2004:49–57
Gruwell ME, Morse GE, Normark BB (2007) Phylogenetic congruence of armored scale insects (Hemiptera: Diaspididae) and their primary endosymbionts from the phylum Bacteroidetes. Mol Phylogen Evol 44(1):267–280
Gruwell ME, Hardy NB, Gullan PJ, Dittmar K (2010) Evolutionary relationships among primary endosymbionts of the mealybug subfamily Phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Appl Environ Microbiol 76(22):7521–7525
Gruwell ME, Flarhety M, Dittmar K (2012) Distribution of the primary endosymbiont (Candidatus Uzinura diaspidicola) within host insects from the scale insect family Diaspididae. Insects 3(1):262–269
Gullan PJ, Cook LG (2007) Phylogeny and higher classification of the scale insects (Hemiptera: Sternorrhyncha: Coccoidea). Zootaxa 1668:413–425
Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Mol Biol Evol 27(3):570–580
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20(14):2317–2319. doi:10.1093/bioinformatics/bth226
Juretschko S, Timmermann G, Schmid M, Schleifer K-H, Pommerening-Röser A, Koops H-P, Wagner M (1998) Combined molecular and conventional analyses of nitrifying bacterium diversity in activated sludge: Nitrosococcus mobilis and Nitrospira-like bacteria as dominant populations. Appl Environ Microbiol 64(8):3042–3051
Kaiwa N, Hosokawa T, Kikuchi Y, Nikoh N, Meng XY, Kimura N, Ito M, Fukatsu T (2010) Primary gut symbiont and secondary, Sodalis-allied symbiont of the scutellerid stinkbug Cantao ocellatus. Appl Environ Microbiol 76(11):3486–3494. doi:10.1128/aem.00421-10
Kane MD, Poulsen LK, Stahl DA (1993) Monitoring the enrichment and isolation of sulfate-reducing bacteria by using oligonucleotide hybridization probes designed from environmentally derived 16S rRNA sequences. Appl Environ Microbiol 59(3):682–686
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Legendre P, Anderson M (1998) Program distpcoa. Département de Sciences Biologiques, Université de Montréal, Montreal
Legendre P, Desdevises Y, Bazin E (2002) A statistical test for host–parasite coevolution. Syst Biol 51(2):217–234
Maidak BL, Cole JR, Lilburn TG, Parker CT Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM (2001) The RDP-II (ribosomal database project). Nucleic Acids Res 29(1):173–174
Matsuura Y, Koga R, Nikoh N, Meng XY, Hanada S, Fukatsu T (2009) Huge symbiotic organs in giant scale insects of the genus Drosicha (Coccoidea: Monophlebidae) harbor flavobacterial and enterobacterial endosymbionts. Zool Sci 26(7):448–456
McClure MS (1990) Impact on host plants. In: Rosen D (ed) Armoured scale insects: their biology, natural enemies and control, vol 4B. Elsevier, Amsterdam, pp 289–291
McCutcheon JP, Moran NA (2007) Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc Natl Acad Sci U S A 104(49):19392–19397
McCutcheon JP, Moran NA (2012) Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10(1):13–26
Morales CF (1990) The taxonomic revision of the New Zealand Margarodidae with studies of the life history and biology of two species. Unpublished Ph.D. Thesis, Imperial College of Science and Technology, University of London, United Kingdom
Morales CF (1991) Fauna of New Zealand: Margarodidae (Insecta: Hemiptera), vol 21. DSIR Plant Protection, Auckland
Moran NA, Tran P, Gerardo NM (2005) Symbiosis and insect diversification: an ancient symbiont of sap-feeding insects from the bacterial phylum Bacteroidetes. Appl Environ Microbiol 71(12):8802–8810
Ovadia Y, Fielder D, Conow C, Libeskind-Hadas R (2011) The cophylogeny reconstruction problem is NP-complete. J Comput Biol 18(1):59–65
Page RDM (2003) Tangled trees: phylogeny, cospeciation, and coevolution. University of Chicago Press, Chicago
Page RDM, Charleston MA (1997) Reconciled trees and incongruent gene and species trees. Math Hierarchies Biol 37:57–70
Park D-S, Suh S-J, Oh H-W, Hebert PDN (2010) Recovery of the mitochondrial COI barcode region in diverse Hexapoda through tRNA-based primers. BMC Genomics 11. doi:10.1186/1471-2164-11-423
Pollard DA, Iyer VN, Moses AM, Eisen MB (2006) Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet 2(10):e173. doi:10.1371/journal.pgen.0020173
Posada D (2009) Selection of models of DNA evolution with jModelTest. Methods Mol Biol 537:93–112
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35(21):7188–7196
Rambaut A (2007) FigTree, a graphical viewer of phylogenetic trees. Available from http://tree.bio.ed.ac.uk/software/figtree. University of Edinburg, UK
Rosenblueth M, Sayavedra L, Sámano-Sánchez H, Roth A, Martínez-Romero E (2012) Evolutionary relationships of flavobacterial and enterobacterial endosymbionts with their scale insect hosts (Hemiptera: Coccoidea). J Evol Biol 25(11):2357–2368. doi:10.1111/j.1420-9101.2012.02611.x
Sabree ZL, Huang CY, Okusu A, Moran NA, Normark BB (2013) The nutrient supplying capabilities of Uzinura, an endosymbiont of armoured scale insects. Environ Microbiol. doi:10.1111/1462-2920.12058
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114–1116
Smit E, Leeflang P, Wernars K (1997) Detection of shifts in microbial community structure and diversity in soil caused by copper contamination using amplified ribosomal DNA restriction analysis. FEMS Microbiol Ecol 23(3):249–261
Stouthamer R, Breeuwer JAJ, Hurst GDD (1999) Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol 53:71–102
Swofford DL (2003) PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods), version 4.0 b10. Sinauer Associates, Sunderland
Tajima F (1993) Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135(2):599–607
Takiya DM, Tran PL, Dietrich CH, Moran NA (2006) Co-cladogenesis spanning three phyla: leafhoppers (Insecta: Hemiptera: Cicadellidae) and their dual bacterial symbionts. Mol Ecol 15(13):4175–4191
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Taylor MW, Schupp PJ, Dahllof I, Kjelleberg S, Steinberg PD (2004) Host specificity in marine sponge associated bacteria, and potential implications for marine microbial diversity. Environ Microbiol 6(2):121–130
Toenshoff ER, Gruber D, Horn M (2012) Co-evolution and symbiont replacement shaped the symbiosis between adelgids (Hemiptera: Adelgidae) and their bacterial symbionts. Environ Microbiol 14(5):1284–1295. doi:10.1111/j.1462-2920.2012.02712.x
Wernegreen JJ (2002) Genome evolution in bacterial endosymbionts of insects. Nat Rev Genet 3(11):850–861
Whiting MF, Carpenter JC, Wheeler QD, Wheeler WC (1997) The Stresiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst Biol:1–68
Wren BW (2002) Deciphering tsetse’s secret partner. Nat Genet 32(3):335–336
Acknowledgments
We would like to thank Sandra Anderson, Robert Brown, Auckland Regional Council and Department of Conservation, New Zealand for facilitating scale insect collections. We are grateful to Dr Bevan Weir for valuable advice on the manuscript and Te Haumihiata from the Maori Language Commission, New Zealand for help with the naming of the B-symbiont. Funding was provided by a University of Auckland International Doctoral Scholarship to M.K.D. and a University of Auckland Faculty Research Development Fund grant (9841 3624443) to J.R.B and M.W.T.
Data Accessibility
DNA sequences: GenBank accessions JX513961 - JX514003, KC447385 - KC447453 and KF697119.
Specimen vouchers: Voucher slides of specimens are accessioned in the New Zealand Arthropod Collection slide collection (institute code: NZAC), Landcare Research, Auckland, New Zealand. Accession numbers are in Table 1.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 47.5 kb)
Rights and permissions
About this article
Cite this article
Dhami, M.K., Buckley, T.R., Beggs, J.R. et al. Primary symbiont of the ancient scale insect family Coelostomidiidae exhibits strict cophylogenetic patterns. Symbiosis 61, 77–91 (2013). https://doi.org/10.1007/s13199-013-0257-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13199-013-0257-8