
Abstract
Antioxidant activities and selected characteristics of gelatin hydrolysates from seabass skin as affected by production processes were investigated. Hydrolysates were prepared using different processes, including hydrolysis during and after gelatin extraction. Samples hydrolysed during gelatin extraction showed a higher degree of hydrolysis (DH) and yield compared with those hydrolysed after gelatin extraction (p < 0.05). All hydrolysates had a creamy yellowish colour. A lower abundance of volatile compounds was found in the hydrolysates produced during gelatin extraction, in comparison with those obtained after gelatin extraction. Hydrolysates prepared during gelatin extraction had higher 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity, ferric reducing antioxidative power (FRAP) and ferrous ion chelating activity (p < 0.05). Following a simulated in vitro gastrointestinal digestion, the DPPH radical scavenging activity and FRAP of the hydrolysates was retained, whilst ferrous ion chelating activity increased. The most appropriate conditions for the generation of antioxidant hydrolysates from seabass skin were identified.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Protein hydrolysates have attracted increasing interest as potential ingredients for various health-promoting functional foods due to their bioactivities (Gómez-Guillén et al. 2011). Collagen and gelatin have been earmarked as a source of biologically active peptides with promising health benefits for nutritional or pharmaceutical applications (Gómez-Guillén et al. 2011). Production of gelatin hydrolysates from fish skins may provide an alternative to meat that is acceptable for Kosher and Halal products and may also serve as a substitute for markets concerned about bovine spongiform encephalopathy (BSE). A number of commercial proteases have been used for the production of gelatin derived hydrolysates and peptides. Alcalase, a commercial protease from a microbial source, has been used in numerous studies investigating gelatin hydrolysis because of its broad specificity as well as the high degree of hydrolysis that can be achieved in a relatively short time under moderate conditions (Benjakul and Morrissey 1997). This enzyme demonstrated extensive proteolytic activity during the hydrolysis of skin gelatin from Alaska pollack, sole and giant squid, producing hydrolysates which exhibited high antioxidant activity (Giménez et al. 2009; Kim et al. 2001).
Seabass (Lates calcarifer) is an economically important fish species in Thailand and other countries, especially tropical and subtropical regions of Asia and the Pacific. During processing or dressing of seabass, skin is generated and considered as a byproduct. Recently, seabass skin has been used to produce collagen and gelatin, as well as hydrolysates with demonstrated bioactivities (Senphan and Benjakul 2014; Sinthusamran et al. 2013). It is well known that protein hydrolysates and peptides can act as free radical scavengers, as well as transition metal chelators and exert antioxidant activities against enzymatic (lipoxygenase-mediated) and non-enzymatic peroxidation of lipids and fats (Sarmadi and Ismail 2010). Gelatin hydrolysates from bigeye snapper skin prepared using a protease derived from fish pyloric caeca exhibited antioxidative activity including DPPH and ABTS radical scavenging activity and ferric reducing antioxidant power (Phanturat et al. 2010). Kittiphattanabawon et al. (2012) reported that gelatin hydrolysates from blacktip shark skin prepared using papaya latex enzyme also showed antioxidant activities in various model systems.
In vitro digestion models are widely used to study the structural changes, digestibility, and release of food components under simulated gastrointestinal conditions (Hur et al. 2011). Stability of antioxidant peptides during gastrointestinal digestion is an important parameter governing bioactivity of a peptide in vivo. When peptides pass through the gastrointestinal tract, they are modified by gastrointestinal proteinases, which could consequently alter antioxidant properties. The effects of an in vitro gastrointestinal digestion on the antioxidant activity of peptides may be used as an indicator of the stability and hence the potential bioactivity of peptides in vivo.
The direct hydrolysis of pretreated skin, a major source of collagen and gelatin, without prior gelatin extraction, can shorten the processing time and production costs. The obtained peptides can serve as a functional supplement in foods or drinks. The present study aimed to investigate the impact of different processing techniques on the selected characteristics and antioxidant activities of hydrolysates from seabass skin. In addition, the impact of a simulated in vitro gastrointestinal digestion on the antioxidant activity of the hydrolysates was evaluated.
Materials and methods
Chemicals
Alcalase (EC 3.4.21.62) (food grade) from Bacillus licheniformis was obtained from Novozyme (Bagsvaerd, Denmark). 2,2′-Azobis(2-amidinopropane) dihydrochloride (AAPH) was from Fluka Chemie (Buchs, Switzerland). 2,4,6-Trinitrobenzenesulphonic acid (TNBS), 2,2-diphenyl-1-picrylhydrazyl (DPPH), 2,4,6-tripyridyltriazine (TPTZ), 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-4′,4″-disulphonic acid sodium salt (ferrozine), ethylenediaminetetraacetic acid (EDTA), 6-hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid (Trolox), porcine pepsin (EC 3.4.23.1, 480 units/mg), glycodeoxycholate, Hank’s balanced salts solution (HBSS), sodium taurocholate, sodium taurodeoxycholate and porcine pancreatin (EC 232-468-9) were procured from Sigma–Aldrich, Inc. (St. Louis, MO, USA). All other chemicals and reagents were from Sigma Chemical Co. (Dublin, Ireland). All solvents used were of HPLC grade.
Collection of seabass skins
Descaled skins of fresh seabass (L. calcarifer) with a weight of 2.5–3 kg were obtained from the Kingfisher Holdings Co., Ltd., Songkhla, Thailand. Skins were kept in a polystyrene box containing ice using a skin:ice ratio 1:2 (w/w) and transported to the Department of Food Technology, Prince of Songkla University, Hat Yai within 1 h. Upon arrival, the remaining meat was removed manually. The skins were washed with cold tap water (1–3 °C). The skins were pooled and used as the composite sample. The samples were placed in polyethylene bags and stored at –20 °C until used, but not longer than 2 months. Prior to gelatin extraction and hydrolysis, the frozen skins were thawed with running water (25–26 °C) until the core temperature reached 8–10 °C. The skins were then cut into small pieces (1.0 × 1.0 cm2) using scissors.
Preparation of seabass skins
Removal of non-collagenous proteins
The skins were soaked in 0.05 M NaOH with a skin:alkali solution ratio of 1:10 (w/v) in order to remove non-collagenous proteins. The mixture was stirred for 3 h at room temperature (28–30 °C) using an overhead stirrer model W20.n (IKA®-Werke GmbH & CO.KG, Stanfen, Germany) at a speed of 300 rpm. The alkaline solution was changed every 1 h for a total of 3 times. Alkali-treated skin was washed with tap water until a neutral or faintly basic pH of wash water was obtained.
Acid pretreatment and defatting
Acid pretreatment and defatting of skin was performed as described by Sae-leaw et al. (2015). Following treatment with alkaline solution, the obtained skins were subjected to swelling process using 0.05 M citric acid at a skin:solution ratio of 1:10 (w/v). The mixture was stirred at room temperature for 2 h. The swollen skin was washed using tap water until wash water became neutral or faintly acidic in pH.
Acid pretreated skins were defatted using 30 % isopropanol with a solid:solvent ratio of 1:10 (w/v) at room temperature for 1 h. The mixture was continuously shaken at 200 rpm. The solvent was then removed and the defatted skin was washed with 10 volumes of tap water to ensure the solvent was completely removed. The defatted skin was then rinsed with distilled water.
Preparation of gelatin hydrolysate
Gelatin hydrolysis was done either during gelatin extraction or after gelatin extraction according to the method of Senphan and Benjakul (2014) with a slight modification. Alcalase was used to hydrolyse gelatin and protease activity was determined as per the method of An et al. (1994). The prepared skins were mixed with distilled water at a ratio of 1:10 (w/v). Prior to hydrolysis, the pH of the mixture was adjusted to 8.0 using 2 M NaOH. For samples hydrolysed during gelatin extraction, Alcalase was added into the mixture at concentrations of 1 or 2 units/g swollen skin dry matter. The mixtures were incubated for 6 h at 55 °C in a water bath with continuous stirring using an overhead stirrer at a speed of 150 rpm. For remaining samples, the gelatins were extracted for 6 h at 55 °C (Sae-leaw and Benjakul 2015) and then hydrolysed with Alcalase at concentrations of 1 or 2 units/g swollen skin dry matter for 3 h at 55 °C. Following hydrolysis, all samples were heated in a boiling water bath for 10 min to inactivate the enzyme. The obtained hydrolysates were centrifuged at 8000×g for 10 min. The supernatants were lyophilised prior to analyses. Gelatin hydrolysates produced during gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin were referred to as ‘P1-1U’ and ‘P1-2U’, respectively. Gelatin hydrolysates obtained after gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin were referred to as ‘P2-1U’ and ‘P2-2U’, respectively.
In vitro gastrointestinal digestion
The simulated in vitro gastrointestinal digestion procedure was performed according to the method of Garrett et al. (1999) with minor modifications. Briefly, 0.4 g of each sample was dissolved in 8 mL Hank’s balanced salts solution (HBSS) and the solution was transferred to an amber bottle. To mimic the gastric phase of human digestion, freshly prepared pepsin (0.04 g/mL in 0.1 N HCl) was added and the pH was adjusted to 2.0 using 0.1 N HCl. The samples were overlaid with nitrogen gas and incubated at 37 °C for 1 h in an orbital shaking water bath (Grant OLS200, Grant Instruments, Cambridge, UK) at 95 rpm. After gastric digestion, the pH was increased to 5.3 using 0.9 M sodium bicarbonate, followed by the addition of 200 μL of freshly prepared bile salts (0.04 g/mL glycodeoxycholate, 0.04 g/mL taurocholate and 0.025 g/mL taurodeoxycholate) and 100 μL of porcine pancreatin (0.08 g/mL). Subsequently, the pH was increased to 7.4 using 1 M NaOH. Samples were overlaid with a layer of nitrogen gas and incubated for a further 2 h at 37 °C in an orbital shaking water bath to mimic the duodenal phase of human digestion. Following intestinal digestion, the digested samples were centrifuged at 14,000×g (Sigma 4K15) for 30 min at 4 °C. The supernatant was collected and filtered through a 0.45 μm membrane filter. Samples were stored at −18 °C until further analysis.
Analyses
Degree of hydrolysis (DH)
DH of the hydrolysates was determined as described by Benjakul and Morrissey (1997). Hydrolysate samples (125 μL) were mixed with 2.0 mL of 0.2 M phosphate buffer (pH 8.2) and 1.0 mL of 0.01 % TNBS solution. The solution was mixed thoroughly and placed in a temperature controlled water bath at 50 °C for 30 min in the dark. The reaction was terminated by adding 2.0 mL of 0.1 M sodium sulphite. The mixture was cooled at room temperature for 15 min. The absorbance was read at 420 nm using a UV-1601 spectrophotometer (Shimadzu, Kyoto, Japan) and α-amino group was expressed in terms of L-leucine. The DH was calculated as follows:
where L is the amount of α-amino groups of hydrolysate sample. L 0 is the amount of α-amino groups in the original swollen skin. L max is the total α-amino groups in the swollen skin obtained after acid hydrolysis (6 M HCl at 100 °C for 24 h).
Yield
The yield of gelatin hydrolysate was calculated based on the dry weight of the swollen skin dried matter by the following equation:
Colour
The colour of the gelatin hydrolysates was measured by a Hunter lab colourimeter (Color Flex, Hunter Lab Inc., Reston, VA, USA). L*, a* and b* indicating lightness/brightness, redness/greenness and yellowness/blueness, respectively, were recorded. The colourimeter was calibrated with a white standard. Total difference in colour (ΔE*) was calculated according to the following equation:
where ΔL*, Δa* and Δb* are the differences between the corresponding colour parameter of the sample and that of white standard (L * = 93.55, a * = − 0.84, b * = 0.37).
Volatile compounds
Volatile compounds in gelatin hydrolysate samples were determined using solid-phase microextraction gas chromatography mass spectrometry (SPME-GC-MS) following the method of Iglesias and Medina (2008) with a slight modification.
To extract volatile compounds, 1 g of sample was mixed with 4 ml of deionised water and stirred continuously to disperse the sample. The mixture was heated at 60 °C in 20 ml headspace vial with an equilibrium time of 10 h. The SPME fibre (50/30 μm DVB/Carboxen™/ PDMS StableFlex™; Supelco, Bellefonte, PA, USA) was conditioned at 270 °C for 15 min before use and then exposed to the headspace. The 20 ml-vial (Agilent Technologies, Palo Alto, CA, USA) containing the sample extract and the volatile compounds was allowed to absorb into the SPME fibre at 60 °C for 1 h. The volatile compounds were then desorbed in the GC injector port for 15 min at 270 °C.
GC–MS analysis was performed in a HP 5890 series II gas chromatography (GC) coupled with HP 5972 mass-selective detector equipped with a splitless injector and coupled with a quadrupole mass detector (Hewlett Packard, Atlanta, GA, USA). Compounds were separated on a HP-Innowax capillary column (Hewlett Packard, Atlanta, GA, USA) (30 m × 0.25 mm ID, with film thickness of 0.25 μm). The GC oven temperature program was: 35 °C for 3 min, followed by an increase of 3 °C/min to 70 °C, then an increase of 10 °C/min to 200 °C, and finally an increase of 15 °C/min to a final temperature of 250 °C and holding for 10 min. Helium was employed as a carrier gas, with a constant flow of 1 ml/min. The injector was operated in the splitless mode and its temperature was set at 270 °C. Transfer line temperature was maintained at 260 °C. The quadrupole mass spectrometer was operated in the electron ionisation (EI) mode and source temperature was set at 250 °C. Initially, full scan mode data was acquired to determine appropriate masses for the later acquisition in scan mode under the following conditions: mass range: 25–500 amu and scan rate: 0.220 s/scan. All the analyses were performed with ionisation energy of 70 eV, filament emission current at 150 μA, and the electron multiplier voltage at 500 V.
Identification of volatile compounds in the samples was done by consulting ChemStation Library Search (Wiley 275.L). Identification of compounds was performed, based on the retention time and mass spectra in comparison with those of standards from ChemStation Library Search (Wiley 275.L). Quantification limits were calculated to a signal-to-noise (S/N) ratio of 10. The identified volatile compounds related to lipid oxidation, including aldehydes, alcohols, ketones, etc., were presented as abundance of each identified compound.
Antioxidant activities
DPPH radical scavenging activity
DPPH radical scavenging activity was determined as described by Brand-Williams et al. (1995) with a slight modification. Sample (100 μL) was mixed with 900 μL of 0.06 mM DPPH in methanol. The sample was mixed vigorously and allowed to stand at room temperature in the dark for 1 h. The absorbance of the resulting solution was measured at 515 nm using a microplate reader (Thermo Scientific Varioskan® Flash Multimode Reader, Fisher Scientific UK Ltd., Leicestershire, UK). The blank was prepared in the same manner except that distilled water was used instead of the sample. The scavenging effect was calculated as follows:
where A is A515 of sample and B is A515 of the blank.
Ferric reducing antioxidant power (FRAP)
The ferric reducing antioxidant power (FRAP) assay was carried out as described by Benzie and Strain (1996). Briefly, 2 mL of working FRAP reagent (0.01 M TPTZ in 0.04 M HCl, 0.02 M FeCl3.6H2O and 0.3 M acetate buffer), prepared fresh daily, was mixed with 1 mL sample. After a 2 h incubation in the dark, absorbance was measured at 593 nm. A standard curve was prepared using Trolox in the range of 20–100 μM. Data were expressed as μmol Trolox equivalents (TE)/g sample.
Ferrous ion-chelating ability
The chelating ability of samples toward ferrous ion (Fe2+) was determined according to the method of Thiansilakul et al. (2007) with a slight modification. Briefly, the samples (250 μL) were mixed with 1 mL of 0.1 M sodium acetate buffer pH 4.9 and 50 μL of 2 mM FeCl2. The reaction was initiated by the addition of 5 mM ferrozine (100 μL). Following 20 min incubation at room temperature, the absorbance was measured at 562 nm using a microplate reader (Thermo Scientific Varioskan® Flash Multimode Reader, Fisher Scientific UK Ltd., Leicestershire, UK). The blank was prepared in the same manner except that distilled water was used instead of ferrozine. The ferrous ion-chelating ability was calculated by the equation:
where A is A562 of sample and B is A562 of the blank.
Oxygen Radical Absorbance Capacity (ORAC)
The ORAC assay was performed as described by Kittiphattanabawon et al. (2012) with some modifications. The samples were dissolved in 75 mM phosphate buffer (pH 7.0) to obtain a final concentration of 0.1 mg/mL. The prepared sample (25 μL) was loaded onto a white polystyrene, nontreated 96-well microplate (Costar Corning Inc., Corning, NY, USA). Only the internal wells of the microplate were used. Fifty μL of 0.04 μM fluorescein dissolved in 75 mM phosphate buffer (pH 7.0) was added to each sample. The loaded microplate was allowed to equilibrate at 37 °C for 20 min in a microplate reader (Thermo Scientific Varioskan® Flash Multimode Reader, Fisher Scientific UK Ltd., Leicestershire, UK). The reaction was initiated by the addition of 100 μL of 221 mM AAPH. The reaction was performed at 37 °C. The fluorescence intensity was measured every 5 min for 90 min with excitation and emission filters of 485 and 535 nm, respectively. The control was prepared in the same manner, except that 75 mM phosphate buffer (pH 7.0) was used instead of the sample. The area under the fluorescence decay curve (AUC) of the samples was calculated by the normalised curves with the following equation:
where f1 is the fluorescence reading at the initiation of the reaction and fn is the last measurement. The net AUC was obtained by subtracting the AUC of the blank from that of a sample or standard. Trolox (0–100 μM) was used as the standard. The ORAC was expressed as μmol Trolox equivalents (TE)/g sample.
Statistical analysis
Experiments were run in triplicate using three different lots of samples. The data were subjected to analysis of variance (ANOVA). Comparison of means was carried out by Tukey’s test. Statistical analysis was performed using the Statistical Package for Social Science (SPSS 11.0 for windows, SPSS Inc., Chicago, IL, USA).
Results and discussion
Degree of hydrolysis (DH)
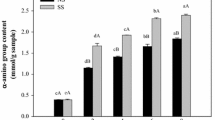
In the present study, seabass skin was hydrolysed by Alcalase for production of antioxidant peptides. Enzymatic hydrolysis of fish proteins generates a mixture of free amino acids, di-, tri-, and oligopeptides, whilst also increasing the solubility of the hydrolysate and improving their functional properties and bioavailability (Bougatef et al. 2012). The extent of protein hydrolysis was estimated by assessing the degree of hydrolysis (DH). DH of gelatin hydrolysates from seabass skin ranged from 36.19 to 38.85 % (Table 1). When swollen seabass skin was hydrolysed using Alcalase at levels of 1 and 2 units/g swollen skin, different DHs were obtained, depending on the process used. A higher DH was observed in P1-1U and P1-2U, which were the hydrolysates produced during gelatin extraction, than in the hydrolysates produced after gelatin extraction (P2-1U and P2-2U). With the same production process, concentration of enzyme (1 or 2 units/g swollen dry skin) did not significantly (p > 0.05) affect DH.
Yield of gelatin hydrolysate
The yields of gelatin hydrolysates prepared from different processes are presented in Table 1. Different yields were obtained for gelatin hydrolysates prepared by different processes (p < 0.05). In general, hydrolysate prepared by gelatin extraction in combination with hydrolysis showed higher yield than those prepared by hydrolysis after gelatin extraction when the same enzyme concentration was used (p < 0.05). The highest yield (67.68 %) was found for P1-2U sample (p < 0.05) and was higher than that of P2-2U (61.76 %). It was found that the yield increased with increasing enzyme levels (p < 0.05). During gelatin extraction at 55 °C, the heat applied destroyed hydrogen bonds stabilizing the triple helix of collagen in the pretreated skin. As a result, the conversion of collagen to gelatin took place. Simultaneously, enzyme, with the optimum temperature of 55 °C, was activated, thereby inducing hydrolysis of the released gelatin. The partially hydrolysed peptides or loosened skin matrix could favor the migration of enzyme to substrate. Also, the exposed substrates were readily available for hydrolysis by protease. During gelatin extraction simultaneous with hydrolysis, peptides released or loosened skin matrix could serve as the preferable substrate rather than the process with gelatin extraction, followed by hydrolysis. This was evidenced by the higher yield when hydrolysis was conducted during gelatin extraction. Thus, production processes affected the yield of gelatin hydrolysate from seabass skin.
Colour of gelatin hydrolysate
The colour of the gelatin hydrolysates obtained from seabass skin was expressed as L*, a* and b* (Table 1). All samples exhibited a creamy yellowish colour. Differences in colour were observed between gelatin hydrolysates from various processes. Generally, hydrolysates produced after gelatin extraction had higher L*- value (lightness) than those hydrolysed during gelatin extraction (p < 0.05). P2-1U exhibited the highest L*- value, whilst P1-2U showed the lowest L*- value (p < 0.05). P1-1U had the lowest a*-value (redness). The lower b*-value (yellowness) was found in sample hydrolysed with the lower level of enzyme. The process used for hydrolysate production did not affect b*-value. Amongst all samples, P1-2U showed the highest ΔE* (total colour difference). This was concomitant with the lowest L*-value. It is likely that at higher concentrations of enzyme, the pigments in the skin were eluted to a greater extent. During hydrolysis, carbonyl compounds (e.g. aldehydes and ketones) produced from lipid oxidation are formed. Those compounds can react with amino groups of free amino acids or peptides via the Maillard reaction (Khantaphant et al. 2011) resulting in a product with a more yellow colour such as that observed in the hydrolysate prepared after gelatin extraction, as indicated by the increased b*-value.
Volatile compounds of gelatin hydrolysate
Selected volatile compounds in seabass skin hydrolysates produced from different processes are shown in Table 2. Aldehydes were found to be the most prolific volatile compounds in gelatin hydrolysates obtained from seabass skin. Aldehydes have been used as the index of lipid oxidation in a number of foods because they possess low threshold values and are the major contributors to the development of off-odour and off-flavour (Ross and Smith 2006). The various aldehyde compounds formed during oxidation included octanal, nonanal, pentanal and hexanal. Hexanal was found as the major aldehyde formed in gelatin hydrolysates (Table 2). It was found that P1-1U and P1-2U had a lower abundance of volatiles in comparison with P2-1U and P2-2U (Table 2). This result suggested that hydrolysis of gelatin during extraction could reduce the hydrolysis time at high temperature, leading to the lower abundance of volatile lipid oxidation products. Iglesias and Medina (2008) reported that propanal, 1-penten-3-ol, 2,3-pentanedione, hexanal and 1-octen-3-ol were the main volatile compounds formed during the storage of Atlantic horse mackerel muscle at 4 °C. Varlet et al. (2006) reported that carbonyl compounds including 4-heptenal, octanal, decanal, and 2,4-decadienal were responsible for the fishy odour of salmon flesh.
Alcohols including 1-penten-3-ol, 1-octen-3-ol, 2-ethylhexanol and 1-hexadecanol were lower in P1-1U than the remaining samples (Table 2). Alcohols are the secondary products produced by the decomposition of hydroperoxides of fatty acids (Ross and Smith 2006). 1-Octen-3-ol is an important contributor to off-flavour due to its low odour threshold and it was reported to be formed from oxidation of arachidonic acid by 12-lipoxygenase (Hsieh and Kinsella 1989). 1-Octen-3-ol contributed to the strong intensity of fishy and rancid off-odours in washed Asian seabass mince containing myoglobin (Thiansilakul et al. 2011). Other volatile compounds were also found in skin hydrolysates. Furans including 2-amylfuran and 5-isopropenyl-3-isopropyl-2,2-dimethyl-2,5-dihydrofuran were detected in all samples.
Generally, secondary oxidation products such as aldehydes and alcohols were lower in P1-1U and P1-2U than in P2-1U and P2-2U indicating that hydrolysis of gelatin during extraction was effective in the reduction of volatile secondary lipid oxidation products, which contribute to the offensive fishy odour in gelatin hydrolysates.
Antioxidant activities of gelatin hydrolysates
The antioxidant activity of protein hydrolysates or peptides can occur via several mechanisms. Therefore, different assays (DPPH, FRAP, ferrous ion chelating ability and ORAC) were performed to assess the antioxidant properties of the gelatin hydrolysates.
DPPH radical scavenging activity
The DPPH radical scavenging assay has been widely used to evaluate antioxidant properties of compounds such as free radical scavengers or hydrogen donors (Klompong et al. 2007). Gelatin hydrolysates from seabass skin showed a dose dependent DPPH scavenging effect ranging from 19 to 79 % (Fig. 1a). At concentrations below 25 mg/mL, no significant difference (p > 0.05) in DPPH radical scavenging activity between samples was found. At higher concentrations, P1-1U had the highest DPPH radical scavenging activity, whilst P2-2U showed the lowest activity. DPPH radical scavenging activity was previously reported in protein hydrolysates prepared from the skin of seabass (Senphan and Benjakul 2014), catfish (Alemán et al. 2011), Alaska pollack (Jia et al. 2010) and bigeye snapper (Phanturat et al. 2010). Increasing quantities of active peptides react with free radicals to convert them to stable products (Batista et al. 2010). At the highest concentration (125 mg/mL), the sample with the highest DPPH scavenging activity (79 %) was P1-1U. The EC50 values (concentration required to inhibit 50 % of DPPH radical) of gelatin hydrolysate were 60, 73, 76 and 87 mg/mL for P1-1U, P1-2U, P2-1U and P2-2U, respectively (data not shown). Samples prepared by hydrolysis after gelatin extraction, showed higher EC50 values and therefore had a lower scavenging activity, than P1-1U and P1-2U (prepared during gelatin extraction). The results indicated that gelatin hydrolysates from seabass skin contain peptides which may act as hydrogen donors to free radicals and thereby have the potential to prevent or retard lipid oxidation via a chain breaking reaction.

DPPH radical scavenging activity (a), FRAP (b) and ferrous ion chelating activity (c) of gelatin hydrolysates from seabass skin prepared using different processes. Bars represent the mean ± standard deviation (n = 3). Different uppercase letters on the bars within the same hydrolysate concentration indicate significant differences (p < 0.05). Different lowercase letters on the bars within the same hydrolysate sample indicate significant differences (p < 0.05), ANOVA followed by Tukey’s. P1-1U and P1-2U represent gelatin hydrolysate produced during gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively. P2-1U and P2-2U represent gelatin hydrolysate produced after gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively
Ferric reducing antioxidant power (FRAP)
The ability of gelatin hydrolysates to reduce Fe3+ to Fe2+ was determined and expressed as FRAP using the well-known antioxidant Trolox as a comparison standard (Fig. 1b). All samples showed FRAP values in the range of 0.35–2.68 μmol TE/g sample. The highest FRAP was obtained in P1-1U at a concentration of 7.5 mg/mL (p < 0.05). No significant differences (p > 0.05) in the FRAP value of P1-1U were observed at different concentrations. Hydrolysates prepared from several fish skins such as bigeye snapper (Phanturat et al. 2010), sole (Giménez et al. 2009) and seabass (Senphan and Benjakul 2014) also exhibited FRAP activity. Processing conditions can greatly influence peptide chain length as well as the exposure of terminal amino groups of products. Greater FRAP values indicate that hydrolysates could donate an electron to free radicals, leading to the prevention or retardation of propagation (Klompong et al. 2007). The results of the present study suggest that gelatin hydrolysates hydrolysed during gelatin extraction possibly contain higher amount of peptides which can donate electrons to free radicals.
Ferrous ion chelating activity
The transition metal ions such as Fe2+ and Cu2+ participate in the formation of free radicals or reactive oxygen species that accelerate lipid oxidation (Sarac 1999), therefore, chelation of transition metal ions could retard or interrupt the oxidation process (Gordon 2001). In comparison with other ions, ferrous ion is a key active species responsible for ROS formation in cells, leading to the increase of lipid oxidation (Huang et al. 2002).
The ferrous ion chelating ability of the gelatin hydrolysates are shown in Fig. 1c. Gelatin hydrolysate samples demonstrated the ability to chelate ferrous ion in a dose dependent manner (p < 0.05). At the same concentration, P1-1U generally showed the highest ferrous ion chelating activity, whilst P2-2U exhibited the lowest activity (p < 0.05). The EC50 values of ferrous ion chelating activity were 57, 84, 112 and 137 mg/mL for P1-1U, P1-2U, P2-1U and P2-2U, respectively (data not shown). The peptides present in the hydrolysates most likely had different metal ion chelating capacity, depending on the amino acid sequences and chain length of peptide fragments. Samaranayaka and Li-Chan (2008) reported that ferrous ion chelating ability ranged from approximately 7 to 46 % for hydrolysates derived from Pacific hake muscle prepared using different hydrolysis processes. Thiansilakul et al. (2007) found a chelating activity of 60 % in round scad protein muscle hydrolysate. Carboxyl and amino groups in the side chains of the acidic (Glx, Asx) and basic (Lys, His, Arg) amino acids are thought to play an important role in chelating metal ions (Saiga et al. 2003). Transition metals, such as Fe, Cu and Co in foods affect both the rate of autoxidation and breakdown of hydroperoxide to volatile compounds. Transition metal ions react very quickly with peroxides by acting as one-electron donors to form alkoxyl radical (Gordon 2001). Skin hydrolysates could act as the secondary antioxidant which could scavenge pro-oxidative metal ions.
Oxygen radical absorbance capacity (ORAC)
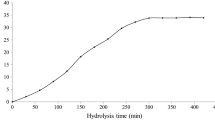
ORAC is the only assay that combines both inhibition time and degree of inhibition into a single quantity (Prior et al. 2005). The protective effect of an antioxidant is calculated from the net integrated area under the fluorescence decay curve (AUC) and reported as Trolox equivalents (Fig. 2a). The fluorescence decay was highest for the control, whilst inhibition was observed in the presence of gelatin hydrolysates. P1-1U showed the highest inhibition toward fluorescence decay (p > 0.05) (Fig. 2a). The ORAC values of the gelatin hydrolysates ranged from 550.25 to 623.88 μmol TE/g sample (Fig. 2b). However, no statistical (p > 0.05) differences in ORAC values were determined between the samples. Kittiphattanabawon et al. (2012) reported that gelatin hydrolysates from blacktip shark skin prepared using papaya latex enzyme showed ORAC in the range of 268.16 to 709.42 μmol TE/g sample.

Fluorescence decay curves of fluorescein (a) and oxygen radical absorbance capacity (ORAC) of gelatin hydrolysates (b). Bars represent the mean ± standard deviation (n = 3). Different letters on the bars indicate significant differences (p < 0.05), ANOVA followed by Tukey’s. P1-1U and P1-2U represent gelatin hydrolysate produced during gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively. P2-1U and P2-2U represent gelatin hydrolysate produced after gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively
Differences in DPPH radical scavenging activity, FRAP, ferrous ion chelating activity and ORAC between gelatin hydrolysates from seabass skin prepared using different processes possibly resulted from differences in the experimental conditions, by which a wide variety of peptides with different modes of actions for inhibiting lipid oxidation were generated during hydrolysis. Changes in size, amount, the exposure of the terminal amino groups of the products obtained and the composition of free amino acids or small peptides affect the antioxidative activity (Thiansilakul et al. 2007). Low molecular weight peptides have generally shown higher antioxidant activity (Qian et al. 2008).
Changes in antioxidant activities of gelatin hydrolysate upon in vitro gastrointestinal digestion
A simulated in vitro gastrointestinal digestion model was used to study the stability of gelatin hydrolysates following incubation with several digestive proteases. In vitro methods for simulating the human digestive tract are being extensively used since they are rapid, safe and do not have the same ethical restrictions as in vivo methods. The antioxidant activity of gelatin hydrolysates, as determined by DPPH scavenging activity, FRAP and ferrous ion chelating activity, following simulated in vitro gastrointestinal digestion are presented in Fig. 3.

Changes in DPPH radical scavenging activity (a), FRAP (b) and ferrous ion chelating activity (c) of gelatin hydrolysates from seabass skin after the simulated in vitro gastrointestinal digestion. Bars represent the mean ± standard deviation (n = 3). Different letters within the same sample indicate significant differences (p < 0.05), ANOVA followed by Tukey’s. P1-1U and P1-2U represent gelatin hydrolysate produced during gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively. P2-1U and P2-2U represent gelatin hydrolysate produced after gelatin extraction with enzyme concentrations of 1 and 2 units/g dry swollen skin, respectively
After simulated gastric digestion, the DPPH radical scavenging activity of all samples slightly decreased (p < 0.05), compared to those of non-digested samples (Fig. 3a). Thereafter, a slight increase was noticeable after intestinal digestion, compared with corresponding gastric digests (p < 0.05). It was noted that no significant differences in DPPH radical scavenging activity of all samples were found between non-digested samples and those obtained after duodenal digestion (p > 0.05). A similar change in antioxidant activity during gastrointestinal digestion of peptides has also been reported recently (Sanjukta et al. 2015). The decrease in activity after pepsin digestion may be due to the degradation of some original peptides and increase in activity was probably due to further formation of peptides with higher antioxidant activity (Sanjukta et al. 2015). The conditions of the gastrointestinal tract, such as digestive enzymes and pH values in the stomach may influence the structures and functions of the peptides. The loss of activity observed could be attributed to the acidic condition (pH 2.0) under gastric phase digestion.
FRAP activity demonstrated a similar trend to DPPH radical scavenging activity following simulated in vitro gastrointestinal digestion. No significant changes in FRAP of all samples were found as a result of the gastric and intestinal digestion, except P1-1U (Fig. 3b). After 1 h of digestion by pepsin, the FRAP of P1-1U decreased slightly but increased following intestinal digestion (p < 0.05). A similar result was observed from collagen peptides from Alaska pollock skin (Guo et al. 2015).
The changes in metal chelating activity of gelatin hydrolysates upon simulated in vitro gastrointestinal digestion are presented in Fig. 3c. The ferrous ion chelating activity of the samples before gastrointestinal digestion ranged from 30.05 to 52.50 % and were significantly decreased (p < 0.05) to 15.87–19.69 % after gastric phase digestion. After duodenal phase digestion, ferrous ion chelating activity increased to 62.61–72.11 % (p < 0.05). In the gastric phase, pepsin may disrupt the structure of peptides and reduce their abilities to bind Fe2+. The decrease in metal chelating activity might also result from the degradation of peptides under high acidic pH in gastric phase. During duodenal phase digestion, pancreatin may cleave the peptides to a greater extent, leading to the release of new antioxidant peptides or modification of the antioxidant peptide sequences. High affinity metal binding groups may became more exposed or be newly formed. Guo et al. (2015) reported that during simulated gastrointestinal digestion of collagen hydrolysate from Alaska pollock skin, different peptides and free amino acids were released by the action of digestive enzymes, which changed the composition of the hydrolysate and the ability of the peptides to bind metal ions. Pepsin shows a high specificity towards the aromatic amino acids e.g. Phe, Tyr, and Trp, and Leu and Glu at the carboxyl side of a peptide bond (Simpson 2000), whilst pancreatin contains many proteases, including endopeptidases (trypsin, α-chymotrypsin, and elastase) and the exopeptidases (carboxypeptidases A and B) with broad specificity (Young et al. 2011). Nalinanon et al. (2011) also found increased antioxidant activity of protein hydrolysate from ornate threadfin bream after being digested in the simulated model.
Conclusions
Production processes affected the selected characteristics and antioxidant activities of seabass skin hydrolysates prepared using Alcalase. Hydrolysis of gelatin during extraction was effective in the reduction of volatile secondary lipid oxidation products. The hydrolysates produced during gelatin extraction demonstrated greater antioxidant activity than those hydrolysates prepared after gelatin had been extracted. DPPH radical scavenging activity and FRAP of seabass skin hydrolysates were stable after in vitro simulated gastrointestinal digestion, whilst metal chelating activity was improved. Therefore, hydrolysate from seabass skin could serve as a potential source of functional food and/or neutraceutical peptides.
References
Alemán A, Giménez B, Gómez-Guillén MC, Montero P (2011) Enzymatic hydrolysis of fish gelatin under high pressure treatment. Int J Food Sci Technol 46:1129–1136
An H, Seymour TA, Wu J, Morrissey MT (1994) Assay systems and characterization of Pacific whiting (Merluccius productus) protease. J Food Sci 59:277–281
Batista I, Ramos C, Coutinho J, Bandarra NM, Nunes ML (2010) Characterization of protein hydrolysates and lipids obtained from black scabbardfish (Aphanopus carbo) by-products and antioxidative activity of the hydrolysates produced. Process Biochem 45:18–24
Benjakul S, Morrissey MT (1997) Protein hydrolysates from Pacific whiting solid wastes. J Agric Food Chem 45:3423–3430
Benzie IFF, Strain JJ (1996) The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239:70–76
Bougatef A, Balti R, Haddar A, Jellouli K, Souissi N, Nasri M (2012) Protein hydrolysates from bluefin tuna (Thunnus thynnus) heads as influenced by the extent of enzymatic hydrolysis. Biotechnol Bioprocess Eng 17:841–852
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. LWT Food Sci Technol 28:25–30
Garrett DA, Failla ML, Sarama RJ (1999) Development of an in vitro digestion method to assess carotenoid bioavailability from meals. J Agric Food Chem 47:4301–4309
Giménez B, Alemán A, Montero P, Gómez-Guillén MC (2009) Antioxidant and functional properties of gelatin hydrolysates obtained from skin of sole and squid. Food Chem 114:976–983
Gómez-Guillén MC, Giménez B, López-Caballero ME, Montero MP (2011) Functional and bioactive properties of collagen and gelatin from alternative sources: a review. Food Hydrocoll 25:1813–1827
Gordon MH (2001) The development of oxidative rancidity in foods. In: Pokorny J, Yanishlieva N, Gordon M (eds) Antioxidants in food. Woodhead Publishing Limited, Cambridge, pp 7–21
Guo L et al (2015) In vitro assessment of the multifunctional bioactive potential of Alaska pollock skin collagen following simulated gastrointestinal digestion. J Sci Food Agric 95:1514–1520
Hsieh RJ, Kinsella JE (1989) Lipoxygenase generation of specific volatile flavor carbonyl compounds in fish tissues. J Agric Food Chem 37:279–286
Huang X, Dai J, Fournier J, Ali AM, Zhang Q, Frenkel K (2002) Ferrous ion autoxidation and its chelation in iron-loaded human liver HepG2 cells. Free Radic Biol Med 32:84–92
Hur SJ, Lim BO, Decker EA, McClements DJ (2011) In vitro human digestion models for food applications. Food Chem 125:1–12
Iglesias J, Medina I (2008) Solid-phase microextraction method for the determination of volatile compounds associated to oxidation of fish muscle. J Chromatogr A 1192:9–16
Jia J, Zhou Y, Lu J, Chen A, Li Y, Zheng G (2010) Enzymatic hydrolysis of Alaska pollack (Theragra chalcogramma) skin and antioxidant activity of the resulting hydrolysate. J Sci Food Agric 90:635–640
Khantaphant S, Benjakul S, Kishimura H (2011) Antioxidative and ACE inhibitory activities of protein hydrolysates from the muscle of brownstripe red snapper prepared using pyloric caeca and commercial proteases. Process Biochem 46:318–327
Kim S-K, Kim Y-T, Byun H-G, Nam K-S, Joo D-S, Shahidi F (2001) Isolation and characterization of antioxidative peptides from gelatin hydrolysate of Alaska pollack skin. J Agric Food Chem 49:1984–1989
Kittiphattanabawon P, Benjakul S, Visessanguan W, Shahidi F (2012) Gelatin hydrolysate from blacktip shark skin prepared using papaya latex enzyme: antioxidant activity and its potential in model systems. Food Chem 135:1118–1126
Klompong V, Benjakul S, Kantachote D, Shahidi F (2007) Antioxidative activity and functional properties of protein hydrolysate of yellow stripe trevally (Selaroides leptolepis) as influenced by the degree of hydrolysis and enzyme type. Food Chem 102:1317–1327
Nalinanon S, Benjakul S, Kishimura H, Shahidi F (2011) Functionalities and antioxidant properties of protein hydrolysates from the muscle of ornate threadfin bream treated with pepsin from skipjack tuna. Food Chem 124:1354–1362
Phanturat P, Benjakul S, Visessanguan W, Roytrakul S (2010) Use of pyloric caeca extract from bigeye snapper (Priacanthus macracanthus) for the production of gelatin hydrolysate with antioxidative activity. LWT Food Sci Technol 43:86–97
Prior RL, Wu X, Schaich K (2005) Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J Agric Food Chem 53:4290–4302
Qian Z-J, Jung W-K, Kim S-K (2008) Free radical scavenging activity of a novel antioxidative peptide purified from hydrolysate of bullfrog skin, Rana catesbeiana Shaw. Bioresour Technol 99:1690–1698
Ross CF, Smith DM (2006) Use of volatiles as indicators of lipid oxidation in muscle foods. Compr Rev Food Sci Food Saf 5:18–25
Sae-leaw T, Benjakul S (2015) Physico-chemical properties and fishy odour of gelatin from seabass (Lates calcarifer) skin stored in ice. Food Biosci 10:59–68
Sae-leaw T, Benjakul S, O’Brien NM (2015) Effect of pretreatments and drying methods on the properties and fishy odor/flavor of gelatin from seabass (Lates calcarifer) skin. Drying Technol. doi:10.1080/07373937.2014.1003071
Saiga A, Tanabe S, Nishimura T (2003) Antioxidant activity of peptides obtained from porcine myofibrillar proteins by protease treatment. J Agric Food Chem 51:3661–3667
Samaranayaka AGP, Li-Chan ECY (2008) Autolysis-assisted production of fish protein hydrolysates with antioxidant properties from Pacific hake (Merluccius productus). Food Chem 107:768–776
Sanjukta S, Rai AK, Muhammed A, Jeyaram K, Talukdar NC (2015) Enhancement of antioxidant properties of two soybean varieties of Sikkim Himalayan region by proteolytic Bacillus subtilis fermentation. J Funct Foods 14:650–658
Sarac AS (1999) Redox polymerization. Prog Polym Sci 24:1149–1204
Sarmadi BH, Ismail A (2010) Antioxidative peptides from food proteins: a review. Peptides 31:1949–1956
Senphan T, Benjakul S (2014) Antioxidative activities of hydrolysates from seabass skin prepared using protease from hepatopancreas of Pacific white shrimp. J Funct Foods 6:147–156
Simpson BK (2000) Digestive proteinases from marine animals. In: Haard NF, Simpson BK (eds) Seafood enzymes: utilization and influence on postharvest seafood quality. Marcel Dekker, Inc., New York, pp 191–214
Sinthusamran S, Benjakul S, Kishimura H (2013) Comparative study on molecular characteristics of acid soluble collagens from skin and swim bladder of seabass (Lates calcarifer). Food Chem 138:2435–2441
Thiansilakul Y, Benjakul S, Shahidi F (2007) Antioxidative activity of protein hydrolysate from round scad muscle using Alcalase and Flavourzyme. J Food Biochem 31:266–287
Thiansilakul Y, Benjakul S, Richards MP (2011) Effect of myoglobin from Eastern little tuna muscle on lipid oxidation of washed Asian seabass mince at different pH conditions. J Food Sci 76:C242–C249
Varlet V, Knockaert C, Prost C, Serot T (2006) Comparison of odor-active volatile compounds of fresh and smoked salmon. J Agric Food Chem 54:3391–3401
Young D, F N, Pasco M, Mine Y (2011) Identification of hen egg yolk-derived phosvitin phosphopeptides and their effects on gene expression profiling against oxidative stress-induced Caco-2 cells. J Agric Food Chem 59:9207–9218
Acknowledgments
This research was supported by a scholarship for an Overseas Thesis Research, Prince of Songkla University Ph.D. Scholarship and the Grant‐in‐Aid for dissertation from Graduate School, Prince of Songkla University, Thailand. School of Food and Nutritional Sciences, University College Cork, Ireland is gratefully acknowledged for the facilities. The Thailand Research Fund (TRF) Distinguished Research Professor Grant was also acknowledged for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sae-leaw, T., O’Callaghan, Y.C., Benjakul, S. et al. Antioxidant activities and selected characteristics of gelatin hydrolysates from seabass (Lates calcarifer) skin as affected by production processes. J Food Sci Technol 53, 197–208 (2016). https://doi.org/10.1007/s13197-015-1989-7
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-015-1989-7