
Abstract
Functional properties and antioxidant activity of pink perch (Nemipterus japonicus) muscle hydrolysed by three different enzymes papain, pepsin and trypsin were studied. The protein hydrolysates produced by trypsin had an excellent solubility (98%) compared to pepsin (77%) and papain hydrolysate (74%). Conversely, the emulsifying activity index (ESI) and foaming abilities were affected by pH. DPPH radical scavenging ability, reducing power and metal chelating activity of protein hydrolysates increased with increase in concentration. Lipid peroxidation was strongly inhibited by 64% by protein hydrolysates produced by trypsin. The results revealed that the functional properties and antioxidant activities of pink perch were greatly affected by the enzymes used.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Free radical mediated oxidation of lipids and formation of secondary lipid peroxidation products is of great concern in food industry. In addition, consuming of oxidative food is thought to cause severe diseases such as cardiovascular diseases, diabetes mellitus, neurological disorders and even Alzheimer ’s disease (Stadtman 2006). Therefore, a great interest has been developed to identify new antioxidant compounds to overcome problems related to lipid peroxidation (Nasirullah and Rakshitha 2009). Many synthetic antioxidants like BHT (butylated hydroxytoluene), BHA (butylated hydroxyanisole) and n-propyl gallate exhibit antioxidant activity stronger than natural antioxidants viz., vit.C and α-tocopherol. However, these synthetic antioxidants are under strict regulation because of their health hazards (Ito et al. 1986).
Several protein hydrolysates derived from food protein have become an important area of research for pharmaceutical, health food and processing/preservation industries (Hagen and Sandnes 2004). Fish protein hydrolysates have been well studied in their production, biochemical and functional properties (Kristinsson and Rasco 2000). The characteristics of the hydrolysates directly affect the functional properties such as solubility, emulsifying and foaming properties (Gbogouri et al. 2004; Kristinsson and Rasco 2000).
Nemipterus japonicus known as pink perch is a major by catch of the shrimp fishery in Indian coastal waters and contributes approximately 3.5% of the total marine landing of India. N. japonicus are widespread throughout the tropical and subtropical Indo-west pacific region. They are valued food fishes in many parts of the world and are taken commercially.
The objective of this study was to evaluate the antioxidant ability of protein hydrolysates obtained from pink perch muscle by enzymatic hydrolysis (trypsin, pepsin and papain), as well as to characterize its functional and nutritional properties.
Materials and methods
Nemipterus japonicus was purchased from Royapuram fishing harbour (Lat 13° 06′ N, Long 80° 18′ E) north of Chennai, India. DPPH (1,1-diphenyl-2-picrylhydrazyl) BHT, and α-tocopherol were purchased from Sigma-Aldrich chemie (Steinheim, Germany). All the other chemicals and solvents were of analytical grade.
Preparation of protein hydrolysate
To produce antioxidant peptides from pink perch muscle, enzymatic hydrolysis was performed using three enzymes namely trypsin (buffer, 0.1 M Na2HPO4–NaH2PO4; pH, 8.8; temperature, 37 °C) pepsin (buffer, glycine –HCl; pH, 2.0; temperature, 37 °C) and papain (buffer, 0.1 M Na2HPO4–NaH2PO4; pH, 6.0; temperature, 37 °C) with their optimal conditions at enzyme /substrate ratio (1/100 w/w). Substrate and enzyme were mixed thoroughly. The mixture was incubated for 4 h at optimal temperature with continuous stirring. The mixture was then heated in a boiling water bath at 100 °C for 10 min to inactivate enzyme and finally lyophilized. The lyophilized hydrolysates were packed in air tight containers and stored at −20 °C until used.
Functional properties of protein hydrolysate
Solubility
Solubility of fish protein hydrolysate was determined over range of pH values from pH 2 to 10. Briefly, 200 mg of protein hydrolysate sample was dispersed in 20 ml of deionized water and pH of the mixture was adjusted 1 N HCl and 1 N NaOH. The mixture was stirred at room temperature for 30 min and centrifuged at 7500 g for 15 min. Protein contents in the supernatant were determined using the Biuret method (Robinson and Hodgen 1940). Total protein content in the sample was determined after solubilization of the sample in 0.5 N NaOH. Protein solubility was calculated as follows:
Emulsifying properties
Emulsifying capacity was determined according to the method of Pearce and Kinsella (1978). Corn oil (10 ml) and 30 ml of 1% protein hydrolysate solution were mixed and the pH of the resulted solution of oil-protein was adjusted to 2, 4, 6, 8 and 10. The mixture was homogenised at a speed of 20,000 rpm for 1 min by using Homogeniser IKA Ultra Turrax T25 (Schott Iberica, S.A., Spain). An aliquot of the emulsion (50 μl) was pipetted from the bottom of the container at 0 and 10 min after homogenization and mixed with 5 ml of 0.1% sodium dodecyl sulphate (SDS) solution. The absorbance of the diluted solution was measured at 500 nm. The absorbances measured immediately (A0) and 10 min (A10) after emulsion formations were used to calculate the emulsifying activity index (EAI) and the emulsion stability index (ESI) as follows:
where as \( \Delta {\text{A = }} {{\text{A}}_{{0 - }}}{{\text{A}}_{{{1}0}}} \) and Δt = 10 min
Foaming properties
Foaming capacity and stability of pink perch protein hydrolysate were determined according to the method of Sathe and Salunkhe (1981). Briefly, 20 ml of 0.5% sample solution were adjusted to pH 2, 4, 6, 8 and 10 followed by homogenization at a speed of 16,000 rpm to incorporate the air for 2 min at room temperature. The whipped sample was immediately transferred into a 25 ml cylinder and the total volume was read after 30 s. The foaming capacity was calculated as,
Where,
- A:
-
is the volume after whipping (ml)
- B:
-
is the volume before whipping (ml).
The whipped sample was allowed to stand at 20 °C for 3 min and the volume of whipped sample was then recorded. Foam stability was calculated as follows:
Where,
- A:
-
volume after standing (ml)
- B:
-
volume before whipping (ml).
Antioxidant assays
DPPH radical scavenging activity
The radical scavenging abilities of the corresponding extracts were measured from bleaching of the purple-colored methanol solution of DPPH. This spectrophotometer assay uses the stable radical DPPH as a reagent (Burits and Bucar 2000). Briefly, 1 ml of various concentrations (0.5–3 mg/ml) of different extracts in ethanol was added to 4 ml of 0.004% methanol solution of DPPH. After 30 min of incubation period at room temperature, the absorbance was read at 517 nm. Inhibition of free radicals by DPPH in percent (I %) was calculated by formula given below,
where A blank is the absorbance of the control reaction (containing all reagents except the test compound), and A sample is the absorbance of the sample.
The control was conducted in the same manner, except that distilled water was used instead of sample. A lower absorbance of the reaction mixture indicated a higher DPPH radical-scavenging activity. Butylated hydroxytoluene (BHT) and α-tocopherol were used as a standard.
Reducing power assay
The reducing ability of the hydrolysates was determined according to the method of Yildirim et al. (2001). An aliquot of 1 ml sample of fish protein hydrolysate at different concentrations (0.5–3.0 mg/ml) was mixed with 2.5 ml of 0.2 M phosphate buffer (pH 6.6) and 2.5 ml of 1% potassium ferricyanide. The mixture was incubated at 50 °C for 30 min, followed by addition of 2.5 ml of 10% (w/v) trichloroacetic acid. The mixture was then centrifuged at 3000 rpm for 10 min. Finally, 2.5 ml of the supernatant solution was mixed with 2.5 ml of distilled water and 0.5 ml of 0.1% (w/v) ferric chloride. After 10 min reaction the absorbance of the resulting solution was measured at 700 nm. Increased absorbance of the reaction mixture indicated increased reducing power.
Metal Chelating activity
The chelating activity on Fe2+ was determined, using the method of Decker and Welch (1990). One milliliter of sample solution with different concentrations 0.5–3 mg/ml) was mixed with 3.7 ml of distilled water. The mixture was then reacted with 0.1 ml of 2 mM FeCl2 and 0.2 ml of 5 mM 3-(2-pyridyl)-5,6-bis(4-phenyl-sulfonic acid)-1,2,4-triazine (ferrozine) for 20 min at room temperature. The absorbance was read at 562 nm. The control was prepared in the same manner except that distilled water was used instead of the sample. Chelating activity (%) was then calculated as follows:
Lipid peroxidation inhibition assay
The oxidation of linoleic acid was conducted as described by Osawa and Namiki (1985). A sample of fish protein hydrolysates were dissolved in 2.5 ml of 50 mM phosphate buffer (pH 7.0), and added to a solution of 2.5 ml of 50 mM of linoleic acid in of 99% ethanol. The total volume was then adjusted to 6.25 ml with distilled water. In the same experiment, sample was replaced with α-tocopherol and butylated hydroxytoluene for comparative purposes. The mixture was incubated in tubes with a screw cap at 40 ± 1 °C for 5 days in a dark room and the degree of oxidation was evaluated by measuring the ferric thiocyanate values according to the method of Mitsuda et al. (1996) A total of 100 ml of the oxidized linoleic acid solution was added to 4.7 ml of 75% ethanol, 0.1 ml of 30% ammonium thiocyanate and 0.1 ml of 20 mM ferrous chloride solution in 3.5% HCl. After stirring for 3 min, the absorbance was measured at 500 nm with α-tocopherol as reference and distilled water as control.
Statistical analysis
All data were expressed as means ± standard deviation of three measurements. Data were analysed by student t–test and all the tests were considered statistically significantly at P < 0.05.
Results and discussion
Functional properties
Solubility
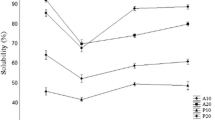
Solubility is one of the most important functional properties of protein hydrolysates (Wilding et al. 1984). The solubility of pink perch protein hydrolysates in different pH ranges 2–10 are shown in Fig. 1. The solubility of protein hydrolysate digested with trypsin showed higher solubility than other two hydrolysates. All the hydrolysates showed significantly (p < 0.05) more than 70% of solubility over wide range of pH which may be due to the degradation of proteins to smaller peptides that lead to more soluble products (Gbogouri et al. 2004). The solubilties of protein hydrolysates were quite low at pH 4 as reported in salmon by products (Gbogouri et al. 2004) and yellow stripe (Klompong et al. 2007). The pH affects the charge on the weakly acidic and basic side chain groups and hydrolysates generally show low solubility at their isoelectric points.

Physio-chemical properties of pink perch protein hydrolysates prepared with different enzymes as affected by pH values (n = 3). Where, EAI denotes emulsifying ability index and ESI is emulsifying stability index
Emulsifying properties
Emulsifying activity index (EAI) and emulsion stability index (ESI) of pink perch at different range of pH 2–10. EAI and ESI of protein hydrolysate were lowest at pH 4 because of decrease in solubility and maximum at pH 6, as given in Fig. 1 respectively. EAI estimates the ability of the protein to aid in the formation and stability of newly created emulsion by giving units of area of interface that is stabilized per unit weight of protein which is determined by the turbidity at 500 nm (Pearce and Kinsella 1978). Protein hydrolysates are surface active substances that promote oil in water emulsion because of their hydrophobic and hydrophilic charges (Gbogouri et al. 2004; Kristinsson and Rasco 2000). EAI and ESI of protein hydrolysate were lowest at pH 4 because of decrease insolubility at this pH 4. EAI and ESI increase with increase in pH that is accompanied to their higher solubility.
EAI and ESI generally increased as pH moved away from pH 4. EAI and ESI also varied with enzymes used, thus suggesting that the sequence and composition of amino acids in peptides of hydrolysates might be different, leading to varying charge of the resulting peptides at a particular pH. This result was also supported by Klompong et al. (2007) in yellow stripe trevally. EAI and ESI of protein hydrolysate produced by trypsin showed maximum EAI and ESI than other two hydrolysates that may be due to their specificity of enzymes which influence emulsifying properties (Gauthier et al. 1993). Emulsion stability is one of the major qualities of emulsion and a number of proteins have been reported to provide desirable emulsifying properties for the preparation of mayonnaise and salad dressing (Song and Damodaran 1987).
Foaming properties
All the three protein hydrolysates showed a lower foaming capacity and foaming stability. As hydrolysis of protein reduces the foaming stability since the microscopic peptide does not have the strength needed to maintain stable foam. The decreasing in foaming of the more extensively hydrolysed sample suggests that lower surfactant activity of smaller polypeptide chains (Mutilangi et al. 1996).
Protein hydrolysates were affected by pH. Foaming capacity tends to decrease at pH 4 and reached maximum at pH 6 and showed slight decrease at alkaline pH 6. Souissi et al. (2007) also indicated that foam capacity in sardinella (Sardinella aurita) decreased with increase in pH. This may be due to the increases in net charge at pH 6 that leads to increase in foaming (Sorgentini and Wagner 2002). The lowest foaming capabilities at pH 4 accomponishes its lower solubility at isoelectric points.
Foam stability depends on the extent of protein-protein interaction within the matrix (Mutilangi et al. 1996). Foam stability was measured at 3 min after whip ability and was found more at pH 6 and less at more acidic and alkaline pH that may be due to result of repulsion of peptides via ionic repulsion.
Among all the three hydrolysates the hydrolysate produced by trypsin showed significantly (p < 0.05)more foaming capacity and foaming stability that might me due to different charge and size of peptides produced by different enzymes as shown in Fig. 1.
Antioxidant assays
DPPH radical scavenging activity
Free radical scavenging ability of three different enzymatic extract of pink perch was evaluated with reduction in absorbance caused by the DPPH radical. DPPH is stable in methanol and shows maximum absorbance at 517 nm.Thus DPPH radicals are used to investigate capabilities of protein hydrolysate.
All the three hydrolysates exhibit the ability to eliminate DPPH radicals at different concentration (0.5–3.0 mg/ml) as shown in Fig. 2. The result indicated the hydrolysates produced by pepsin significantly (p < 0.05) exhibited the highest scavenging activity (48% at 3 mg/ml) compared to other hydrolysates and was less with respect to commercial antioxidant α-tocopherol and BHT (64% and 57%) . Jun et al. (2004) also reported that yellow fin sole hydrolysate prepared from pepsin showed highest DPPH scavenging activity than those produced by other hydrolysates. Further, Sun et al. (2011) also proved that pepsin hydrolysate of porcine haemoglobin showed maximum antioxidant activity than other hydrolysated derived from various proteases. Therefore, the results reveal that pink perch hydrolysates contain electron donors and could react with free radicals to convert into more stable products and terminate the radical chain reaction.

DPPH radical scavenging activity, reducing ability and metal chelating activity of pink perch muscle protein hydrolysates at different concentrations (n = 3)
Reducing ability
The reducing power is used to evaluate the ability to donate electron (Yildirim et al. 2001). The ability of protein hydrolysates to reduce Fe3+ to Fe2+ was studied. Reducing power was concentration dependent and increased with increase in concentration. Earlier studies also reported that the reducing power increased with increasing amount of sample (Zhu et al. 2006). Furthermore, reducing power of pink perch protein hydrolysate varied with the enzymes used for hydrolysis. Klompong et al. (2007) also showed that reducing ability of yellow stripe trevally (Selaroides leptolepis) varied with the enzymes used.
The reducing ability of the different hydrolysate was compared with commercial antioxidant i.e. α-tocopherol and BHT. The higher activity was found in the protein hydrolysate produced by pepsin followed by papain as shown in Fig. 2. Hence, from results it appears that pink perch muscle hydrolysates could be excellent electron donors to free radicals.
Metal chelating
The chelating of ferrous ions was used to determine the ability of hydrolysates in metal chelating activity. Transition metal ions such as Fe2+ and Cu2+ can catalyze the generation of reactive oxygen species which oxidizes unsaturated lipids. The chelating activity of peptides in hydrolysate could decrease lipid oxidation. The chelating activity increased with increase in concentration was earlier reported of yellow stripe trevally (Selaroides leptolepis) (Klompong et al. 2007). Samaranayaka and Li-Chan (2008) reported a chelating ability ranging from approximately 7% to 46% for hydrolysates derived from pacific hake muscle by different hydrolysis procedures, at 5 mg/ml assay concentration. Similarly, Thiansilakul et al. (2007) found a chelating activity of 60% in round scad protein muscle hydrolysate, although the assay concentration was not specified. It was also revealed that metal chelating activity of pink perch was influenced by the enzyme used. Protein hydrolysate produced by pepsin exhibited strong metal chelating activity at 3 mg/ml than other hydrolysates (Fig. 2). However, its metal chelating ability was significantly lower than that of EDTA which has strongest metal chelating ability of 90% at 3 mg/ml.
Lipid peroxidation inhibition assay
In this study, we found the oxidation of linoleic acid was markedly inhibited by hydrolysates derived from pink perch muscle protein with different proteases and was compared to α-tocopherol as shown in Fig. 3. To evaluate lipid peroxidation inhibitory activity, a well known PUFA, linoleic acid was incubated to auto-oxidize in a water/ethanol emulsion in a dark room at 40 ± 1 °C. In this model system, peroxyl (ROO-) and alkoxyl (RO-) radicals, derived from the pre-existing lipid peroxide, were employed directly to initiate lipid peroxidation in the emulsified linoleic acid system. Cheng et al. (2003) reported that phenolic compounds afforded their protective actions in lipid peroxidation by scavenging the lipid derived radicals (R- , RO- or ROO-) to stop the chain reactions in a heterogeneous lipid phase. The hydrolysates resulting from various enzymes, effectively inhibited lipid peroxidation in linoleic acid emulsion system up to the 6 days, and the activity was similar to that of α-tocopherol and the highest antioxidant activity was observed in hydrolysate formed by trypsin which exhibited significantly (p < 0.05) about 64% inhibition of linoleic acid which was too close to α-tocopherol. The control showed the highest absorbance value indicating the highest degree of oxidation. All the hydrolysates showed more than 55% of inhibition of lipid peroxidation as was also seen in Alaska Pollack (Theragra chalcogramma) (Je et al., 2005)

Lipid peroxidation inhibition activity of pink perch muscle protein hydrolysates. Lower absorbance at 500 nm represents higher lipid peroxidation inhibition. Each value represents mean of three different independent experiments ± SD
Conclusions
Functional properties like solubility, emulsifying activity index and foaming abilities of hydrolysates of pink perch (Nemipterus japonicus) varied with enzyme used. Furthermore functional properties were also affected by the pH.
Antioxidant activities varied with respect to enzyme used. Therefore protein hydrolysates of pink perch can serve as good source of antioxidant peptides with essential amino acids and finds its application in food industry, health benefit products or pharmaceuticals. However, furthermore studies are needed with regard to in vivo antioxidant studies.
References
Burits M, Bucar F (2000) Antioxidant activity of Nigella sativa essential oil. Phytother Res 14:323–328
Cheng Z, Ren J, Li Y, Chang W, Chen Z (2003) Establishment of a quantitative structure-activity relationship model for evaluating and predicting the protective potentials of phenolic antioxidants on lipid peroxidation. J Pharm Sci 92:475–84
Decker EA, Welch B (1990) Role of ferritin as a lipid oxidation catalyst in muscle food. J Agric Food Chem 38:674–677
Gauthier SF, Paquin P, Pouliot Y, Turgeon S (1993) Surface activity and related functional properties of peptides obtained from whey proteins. J Dairy Sci 76:321–328
Gbogouri GA, Linder M, Fanni J, Parmentier M (2004) Influence of hydrolysis degree on the functional properties of salmon by product hydrolysates. J Food Sci 69:615–622
Hagen H, Sandnes K (2004) Process for improvement of meat quality in fish, protein hydrolysate and method of producing a protein hydrolysate. International Patent No. WO 2004071202.
Ito N, Hirose M, Fukushima S, Tsuda H, Shirai T, Tatematsu M (1986) Studies on antioxidants: Their carcinogenic and modifying effects on chemical carcinogenesis. Food Chem Toxicol 24:1071–1082
Je JY, Park PJ, Kim SK (2005) Antioxidant activity of peptide isolated from Alaska Pollack (Theragra chalcogramma) frame protein hydrolysate. Food Res Inter 38:45–50
Jun SY, Park PJ, Jung WK, Kim SK (2004) Purification and characterization of an antioxidative peptide from enzymatic hydrolysate of yellowfin sole (Limanda aspera) frame protein. Eur Food Res Technol 219:20–26
Klompong V, Benjakul S, Kantachote D, Shahidi F (2007) Antioxidative activity and functional properties of protein hydrolysate of yellow stripe trevally (Selaroides leptolepis) as influenced by the degree of hydrolysis and enzyme type. Food Chem 102:1317–1327
Kristinsson HG, Rasco BA (2000) Fish protein hydrolysates: production, biochemical, and functional properties. Crit Rev Food Sci Nutr 40:43–81
Mitsuda HK, Yasumoto K, Iwami E (1996) Antioxidative action of indole compounds during the autoxidation of linoleic acid. Eiyoto Shokuryo 19:210–214
Mutilangi WAM, Panyam D, Kilara A (1996) Functional properties of hydrolysates from proteolysis of heat-denatured whey protein isolate. J Food Sci 61:270–274
Nasirullah JT, Rakshitha D (2009) Isolation and antioxidant efficacy of nutraceutical concentrates from sesame and flax seed oils. J Food Sci Technol 46:66–69
Osawa T, Namiki M (1985) Natural antioxidant isolated from eucalyptus leaf waxes. J Agric Food Chem 33:777–780
Pearce KN, Kinsella JE (1978) Emulsifying properties of proteins: evaluation of a turbidimetric technique. J Agric Food Chem 26:716–723
Robinson HW, Hodgen CG (1940) The biuret reaction in the determination of serum protein I A study of the condition necessary for the production of the stable color which bears a quantitative relationship to the protein concentration. J Biol Che 135:707–725
Samaranayaka AGP, Li-Chan ECY (2008) Autolysis-assisted production of fish protein hydrolysates with antioxidant properties forms Pacific hake (Merluccius productus). Food Chem 107:768–776
Sathe SK, Salunkhe DK (1981) Functional properties of the Great Northern bean (Phaseolus vulgaris L.) protei:s: emulsion, foaming, viscosity and gelation properties. J Food Sci 46:71–74
Song KB, Damodaran S (1987) Structure-function relationship of proteins: adsorption and structural intermediates of bovine serum albumin at the air/water interface. J Agric Food Chem 35:236–241
Sorgentini DA, Wagner JR (2002) Comparative study of foaming properties of whey and isolate soy bean proteins. Food Res Inter 35:721–729
Souissi N, Bougatef A, Triki-Ellouz Y, Nasri M (2007) Biochemical and functional properties of sardinella (Sardinella aurita) By-Product Hydrolysates. Food Technol Biotech 45:187–194
Stadtman ER (2006) Protein oxidation and aging. Free Rad Res 40:1250–1258
Sun Q, Shen H, Luo Y (2011) Antioxidant activity of hydrolysates and peptide fractions derived from porcine hemoglobin. J Food Sci Technol 48:53–60
Thiansilakul Y, Benjakul S, Shahidi F (2007) Compositions, functional properties and antioxidative activity of protein hydrolysates prepared from round scad (Decapterus maruadsi). Food Chem 10:1385–1394
Wilding P, Lilliford PJ, Regenstain JM (1984) Functional properties of proteins in food. J Chem Technol Biotech 34:182–189
Yildirim A, Mavi A, Kara AA (2001) Determination of antioxidant and antimicrobial activities of Rumex crispus L. extracts. J Agric Food Chem 49:4083–4089
Zhu K, Zhou H, Qian H (2006) Antioxidant and free radical-scavenging activities of wheat germ protein hydrolysates (WGPH) prepared with alcalase. Process Biochem 41:1296–1302
Acknowledgment
We thank Dr. K. Ramasamy, Dean, School of Bioengineering, SRM University, for his support throughout the project. We would like to extend our acknowledgment to the management, SRM University for providing the facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Naqash, S.Y., Nazeer, R.A. Antioxidant and functional properties of protein hydrolysates from pink perch (Nemipterus japonicus) muscle. J Food Sci Technol 50, 972–978 (2013). https://doi.org/10.1007/s13197-011-0416-y
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-011-0416-y