
Abstract
Metazoa comprises 35–40 phyla that include some 1.3 million described species. Phylogenetic analyses of metazoan interrelationships have progressed in the past two decades from those based on morphology and/or targeted-gene approaches using single and then multiple loci to the more recent phylogenomic approaches that use hundreds or thousands of genes from genome and transcriptome sequencing projects. A stable core of the tree for bilaterian animals is now at hand, and instability and conflict are becoming restricted to a key set of important but contentious relationships. Acoelomorph flatworms (Acoela + Nemertodermatida) and Xenoturbella are sister groups. The position of this clade remains controversial, with different analyses supporting either a sister-group relation to other bilaterians (=Nephrozoa, composed of Protostomia and Deuterostomia) or membership in Deuterostomia. The main clades of deuterostomes (Ambulacraria and Chordata) and protostomes (Ecdysozoa and Spiralia) are recovered in numerous analyses based on varied molecular samples, and also receive anatomical and developmental support. Outstanding issues in protostome phylogenetics are the position of Chaetognatha within the protostome clade, and the monophyly of a group of spiralians collectively named Platyzoa. In contrast to the broad consensus over key questions in bilaterian phylogeny, the relationships of the five main metazoan lineages—Porifera, Ctenophora, Placozoa, Cnidaria and Bilateria—remain subject to conflicting topologies according to different taxonomic samples and analytical approaches. Whether deep bilaterian divergences such as the split between protostome and deuterostome clades date to the Cryogenian or Ediacaran (and, thus, the extent to which the pre-Cambrian fossil record is incomplete) is sensitive to dating methodology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ever since Ernst Haeckel published his artistically illustrated phylogenetic trees, scientists have worked towards understanding the evolution of multicellular animals and the relationships between their main lineages. Although not based on explicit methodology, Haeckel’s (1866) trees are considered hypotheses of relationships and some of these, e.g. the common origin of bilaterian animals, still find support today, whereas numerous others have been falsified by morphological reinterpretations, novel kinds of data derived from new technologies (e.g. electron microscopy, DNA sequencing) and ever-advancing analytical methods.
The path to present-day insights has been shaped by thousands of scientists who have contributed data, methodological developments, and philosophical insights. To mention every single contribution would be impossible in this review, but a few major key events should be addressed briefly because they have been highly significant for our current knowledge. An important philosophical contribution was made by the entomologist Willi Hennig, who defined the outline for modern phylogenetic systematics. Before the ideas of Hennig (1950, 1965, 1966) became established, phylogenetic studies had suffered from several shortcomings, notably the failure to distinguish between monophyletic and paraphyletic groups. Hennig stressed the importance of identifying apomorphic character states and the recognition of clades (monophyletic groups) as the only systematically valid groups. A second major advance was made in the late 1960s and early 1970s with the introduction of explicit numerical methodologies for phylogenetic analysis that used parsimony, likelihood, and other optimality criteria as an arbiter for choosing between competing hypotheses (Farris 1970; Farris et al. 1970; Felsenstein 1973; Kluge and Farris 1969). Up to that point the ideas of Hennig had been adopted by many systematists, but the complexity and number of potential phylogenetically informative characters often forced scientists to base their hypotheses on a few selected characters—e.g. larval ciliary bands, excretory systems, or embryology in the case of deep metazoan relationships—leaving out much other important information and carrying the risk of producing biased results based on homoplasies. Computerized phylogenetic analysis allowed the researcher to handle much larger morphological datasets and analyze the information in a more objective and thorough manner. Hence, within a relatively short period, the computer and phylogenetic software became standard tools for systematists and prompted the publication of numerous morphology-based hypotheses on animal evolution (e.g. Backeljau et al. 1993; Eernisse et al. 1992; Glenner et al. 2004; Nielsen et al. 1995; Schram 1991; Schram and Ellis 1994; Sørensen et al. 2000; Wallace et al. 1996). Other important developments involved refinements in interpretations of homologies (see, for example, the work by R. A. Jenner for critical discussion of characters and character codings; e.g. Jenner 2001, 2004a, 2004b) and the use of species, instead of supraspecific taxa coded as inferred groundplans, for higher-level phylogenetic studies (Prendini 2001).
The most ground-shaking innovation in modern phylogenetics was beyond any doubt the introduction of molecular sequence data. The first attempts to understand metazoan phylogeny through inference of molecular sequences were made in the late 1980s (e.g. Field et al. 1988; Lake 1990; Raff et al. 1989). The molecular approach experienced a substantial breakthrough with the contributions of several authors who redefined the major splits in animal phylogeny, largely using nuclear ribosomal genes (e.g. Aguinaldo et al. 1997; Carranza et al. 1997; Halanych et al. 1995; Winnepenninckx et al. 1995a, b), and convincingly demonstrated the potential for molecular sequence data to recover deep divergences in the animal kingdom.
Since the turn of the millennium, the use of molecular sequence data in phylogenetics has developed rapidly. From using a single selected locus, alone or in combination with morphological data (e.g. Giribet et al. 2000; Giribet and Ribera 1998; Glenner et al. 2004; Peterson and Eernisse 2001; Zrzavý et al. 1998), researchers switched to multilocus approaches (e.g. Giribet 2003; Giribet et al. 2004; Mallatt et al. 2004, 2010; Mallatt and Giribet 2006; Mallatt and Winchell 2002; Paps et al. 2009a; Sørensen et al. 2008; Wallberg et al. 2007), and subsequently to phylogenomic approaches with inclusion of the mitochondrial genomes, complete genomes, or partial transcriptomes (expressed sequence tags, ESTs) (Baguñà et al. 2008; Bourlat et al. 2006; Copley et al. 2004; Dellaporta et al. 2006; Delsuc et al. 2006; Dopazo and Dopazo 2005; Dunn et al. 2008; Hejnol et al. 2009; Lartillot and Philippe 2008; Marlétaz et al. 2006; Matus et al. 2006; Philippe et al. 2004, 2005, 2007; Roeding et al. 2007; Schierwater et al. 2009; Siddall 2009; Webster et al. 2006). Analyses of sequence data have been joined as well by phylogenetic approaches that use rare genomic changes as characters, although these apply mostly to vertebrate genomes. The use of microRNAs, small non-coding RNAs analyzed as absence/presence characters, has also been incorporated into the molecular toolkit in recent years (Rota-Stabelli et al. 2010a; Sempere et al. 2006, 2007; Sperling et al. 2009a, b; Wheeler et al. 2009).
Consequently, myriad phylogenetic hypotheses have been published in the past decade alone, and even the most dedicated systematic researcher may from time to time fail to see the consensus in this deep forest of phylogenetic trees. In the present contribution we review some of the most recent phylogenetic analyses of higher-level metazoan relationships, with emphasis on the newest developments, mostly based on phylogenomic studies. By pointing out congruence and discrepancies between various hypotheses we explore the current consensus in metazoan phylogeny (summarized in Fig. 1), and discuss our present knowledge in relation to morphological and developmental character evolution. We also appraise the merits and limitations of several alternative molecular phylogenetic approaches that are currently used to address the vexing question of metazoan interrelationships.

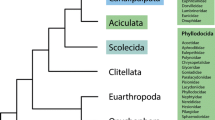
Summary of relationships within Metazoa. Nodes labelled with circled letters (taxon-name abbreviations) have received broad consensus
Results and discussion
Metazoans and phylogenomics
Metazoa (animals) is a monophyletic group of heterotrophic organisms. Apomorphies include special glycoproteins in the form of collagens (Ax 1996), protein kinase C for cell signaling (Gamulin et al. 2000), use of RFamide and acetylcholine (Nickel 2010) as neurotransmitters, and features of gametogenesis (Ax 1996). Metazoa includes approximately 1.3 million described living species (the estimated number of undescribed species ranges from 10 to 200 million) in 35–40 phyla depending on the classification followed, but most recent ones have subsumed many formerly recognized phyla, such as Echiura, Pogonophora, Sipuncula and Vestimentifera into Annelida, Acanthocephala into Rotifera, and even Phoronida into Brachiopoda. For the purpose of this article, we consider the following 36 phyla: Ctenophora, Porifera (as Calcarea, Silicea and Homoscleromorpha in Fig. 1), Cnidaria, Placozoa, Acoela, Nemertodermatida, Xenoturbellida, Echinodermata, Hemichordata, Cephalochordata, Urochordata, Craniata (including Vertebrata), Loricifera, Kinorhyncha, Priapulida, Nematoda, Nematomorpha, Tardigrada, Onychophora, Arthropoda, Annelida, Mollusca, Brachiopoda, Phoronida, Nemertea, Chaetognatha, Bryozoa, Entoprocta, Cycliophora, Platyhelminthes, Gastrotricha, Gnathostomulida, Micrognathozoa, Rotifera, Rhombozoa, and Orthonectida.
The so-called ‘new animal phylogeny’ (Adoutte et al. 2000; Giribet et al. 2007; Halanych 2004), largely driven by molecular data, supports the monophyly of Bilateria, the existence of a large clade of moulting protostome animals named Ecdysozoa (Aguinaldo et al. 1997; Giribet 2003; Schmidt-Rhaesa et al. 1998; Telford et al. 2008), and the presence of a clade that contains the spiralian phyla (Spiralia or Lophotrochozoa; Giribet 2008; Giribet et al. 2000, 2009; Halanych 2004; Halanych et al. 1995). Two main bilaterian clades, Protostomia and Deuterostomia (Fig. 1), are well supported in phylogenomic analyses with broad taxon sampling (Dunn et al. 2008; Hejnol et al. 2009; Philippe et al. 2009).
Although the major clades of animals based on molecular analyses have now been recognized for nearly a decade, resolving the more detailed relationships among the phyla within each major clade with high levels of support and/or stability has been difficult using target-gene approaches that rely on directed sequencing of PCR products. This lack of clear resolution has been presented frequently in the literature as an intractable signature of the Cambrian explosion, even after using sequence data from 50 genes (Rokas et al. 2005). Alternatively, the lack of resolution has been suspected to reflect the limits of the available data, which were restricted in terms of taxon number, breadth of taxon sampling, and the number and types of genes sequenced. This has led to the broadly held conclusion that increased sampling, across both genes and taxa, is a prerequisite for improved resolution of the tree of life (Sanderson 2008). Phylogenomic methods—the construction of species trees using many genes—have rapidly expanded in the past few years to meet this challenge.
Deep gene sampling across many broadly sampled taxa required new technical approaches. Directed sequencing of PCR fragments works well across a broad number of taxa, but does not scale well to large numbers of genes. Genome sequencing is the only way to generate a nearly comprehensive set of gene predictions for an organism, but remains too expensive to scale across broadly sampled taxa. Expressed Sequence Tags (ESTs), randomly acquired sequence fragments of expressed genes obtained from complimentary DNA libraries, have proved to be a productive intermediate solution. Since the first animal phylogenomic analyses using EST data, relationships have been resolved for several groups of phyla (Bleidorn et al. 2009; Bourlat et al. 2006; Delsuc et al. 2005, 2006; Dunn et al. 2008; Egger et al. 2009; Hausdorf et al. 2007, 2010; Hejnol et al. 2009; Helmkampf et al. 2008a, b; Marlétaz et al. 2006; Matus et al. 2006; Philippe et al. 2005, 2007, 2009; Roeding et al. 2007, 2009; Struck and Fisse 2008; Webster et al. 2006; Witek et al. 2009).
Three important limitations plagued the first animal phylogenomic studies. First, they added data for only a small number of taxa (typically 1–3) at a time, so sampling was heavily biased towards organisms that were already well represented in public archives (e.g. vertebrates, insects, nematodes, and molluscs). This left many key groups entirely unrepresented, making it impossible to rigorously test the positions of the added taxa. Second, these studies did not fully leverage the potential of ESTs to inform gene selection, relying instead on manually curated gene lists, particular groups of proteins selected a priori, or gene lists from previous studies. In contrast to targeted-gene studies based on PCR amplification of particular genes, gene selection in EST studies can be part of the data analysis process rather than a facet of project design (Fig. 2). Gene selection after data acquisition has several potential advantages, including a more detailed assessment of orthology, the ability for explicit evaluation of informativeness (Townsend 2007), and improved scalability. Third, many phylogenomic studies have not yet released to public databases all the data on which they are based. In these cases, only the subsets of genes considered in the final analyses are released, which makes it impossible for other investigators to include these data in matrices that use different sets of genes.

In order to overcome these limitations of prior animal EST studies, Dunn et al. (2008) generated a more broadly sampled phylogenomic analysis of animals. This study contributed new EST data for 29 animal species that were explicitly selected to be as complimentary as possible to existing data. New approaches were developed for orthology identification and gene selection, producing a matrix of 150 genes with 50.9% occupancy across 77 taxa. This study reinforced several previously identified clades that split deeply in the animal tree (including Protostomia, Ecdysozoa, and Spiralia), and provided new insight into a number of long-standing issues for which there was strong conflicting support in earlier studies with fewer data. A follow-up study (Hejnol et al. 2009) added EST data for additional representatives of key groups, for which phylogenomic data had been unavailable (Cycliophora and Nemertodermatida) or which had been the most unstable taxa in the previous study (Acoela); furthermore, gene predictions from recently completed genomes for a placozoan (Srivastava et al. 2008) and a gastropod mollusc were added. A scalable gene selection strategy was developed that relied on explicit criteria and was fully automated. This approach allowed an expansion of gene sampling to 1487 genes. Encouragingly, the most unstable taxon in the previous analysis (Dunn et al. 2008), Acoela, was placed along with other acoelomorphs as sister group to the remaining Bilateria, with strong support following increased sampling and improved gene selection methods.
Now that it is possible to automate matrix construction from large genomic and transcriptome datasets, one of the most pressing analytical challenges in phylogenomic analyses is to develop new methods for assessing which character sets support which phylogenetic hypotheses. Phylogenetic inference is too computationally intensive to assess a large number of ad hoc combinations of characters and taxa (e.g. removing different gene sets to see if they are in conflict with a particular topology); thus, approaches that can further dissect the signal from a smaller number of large analyses will be increasingly important.
The basal metazoan lineages
Five major extant lineages of animals result from the deepest splits in the animal tree of life: Bilateria, Cnidaria, Ctenophora, Placozoa, and Porifera. It is widely believed that each of these groups is monophyletic; the only exception in recent literature has been the proposition that Porifera is paraphyletic, giving rise to all other animals (Sperling et al. 2009a, 2010), but this has been contradicted by much larger samples of genes that defend sponge monophyly (Philippe et al. 2009). The topology and rooting of this five-taxon tree are still in dispute; the different studies either give unresolved or strongly conflicting results, or lack broad enough taxon sampling to address the issue. These are some of the most fundamental questions in metazoan phylogeny, and our ability to reconstruct the most recent common ancestor of animals, determine which characters were present before animals radiated, and establish the sequence of character changes that resulted in the differences between these groups lies in the balance.
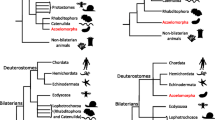
From the 105 possible rooted bifurcating trees for five taxa, only a few possibilities have been proposed (see Fig. 3 for some recent examples). Improved taxon sampling within the five extant lineages of Metazoa and, perhaps even more importantly, for the outgroup taxa is necessary to resolve this issue with more support. Few published phylogenomic analyses include representatives of all five metazoan groups, which greatly limits comparison across existing studies. The traditional view held for more than 100 years, that Porifera is sister group to all other animals (Fig. 3a), had been based on their lack of tissue organization, lack of a nervous system, and the similarity of choanocytes to choanoflagellates (e.g. Nielsen 2001). The placozoan genome paper included a phylogenomic analysis of Placozoa, Porifera, Cnidaria, and Bilateria, and was consistent with the classical view of sponges as the sister group to all other metazoans (Srivastava et al. 2008). Ctenophores, however, were absent from this analysis.

Some postulated relationships among the five main metazoan clades. a Traditional hypothesis based on morphology, with sponges as sister group to other metazoans (e.g. Nielsen 2001). b Hypothesis based on phylogenomic analyses, with ctenophores as sister group to all other metazoans (Dunn et al. 2008; Hejnol et al. 2009). c Hypothesis by Schierwater et al. (2009), with Bilateria as sister group to a clade that contains placozoans as sister to sponges, cnidarians and ctenophores. d Hypothesis with monophyletic Coelenterata (Philippe et al. 2009). e Hypothesis with monophyletic Eumetazoa but with Ctenophora as sister to all other eumetazoans (Pick et al. 2010)
An analysis of mitochondrial genomes, which again did not include Ctenophora, found that Bilateria was sister group to Porifera, Cnidaria and Placozoa (Dellaporta et al. 2006), consistent with the topology of Fig. 3c. The authors claimed at the time, however, that this was an artefact introduced by Bilateria. When Bilateria was excluded, they found that the remaining metazoan tree rooted along the stem of Placozoa—the only molecular analysis to support Placozoa as sister group to other animals. The same authors subsequently published an analysis that included ctenophores, a similar mitochondrial dataset, nuclear genes, and a small morphological matrix (Schierwater et al. 2009), concluding that the placozoan rooting was spurious, and that Bilateria is sister group to the remaining metazoans (Fig. 3c). The placement of Ctenophora as sister group to the remaining metazoans was, however, statistically indistinguishable from the bilaterian rooting in that study (see their Table 1).
The analyses of Dunn et al. (2008) and Hejnol et al. (2009), including representatives of all five groups, placed Ctenophora as sister group to the remaining animals (Fig. 3b). Sampling within these lineages varied greatly, and several important poriferan lineages in particular were not represented. These analyses only included representatives of Demospongiae (ESTs for Suberites domuncula and whole genome sequence data of Amphimedon queenslandica) and Homoscleromorpha (Oscarella lobularis), but no data from the two other sponge groups, Calcarea and Hexactinellida. Two (Dunn et al. 2008) and three (Hejnol et al. 2009) species of ctenophores were included, respectively, but uncertainty regarding the internal ctenophore phylogeny makes it unclear if the basal ctenophore node was bracketed. In spite of these obvious limitations regarding taxon sampling (Pick et al. 2010), the placement of ctenophores was well supported and consistent across all analyses of these matrices, including different subsets of taxa and genes. The inclusion of Placozoa in the analysis of Hejnol et al. (2009), missing in the Dunn et al. (2008) dataset, did not alter the placement of Ctenophora as sister group to all other Metazoa.
More recent EST analyses that also included all five groups of metazoans included much improved sponge sampling (Philippe et al. 2009; Pick et al. 2010). These new data include ESTs for all major groups of sponges: Homoscleromorpha (O. lobularis), Calcarea (Sycon raphanus, Leucetta chagosensis), Demospongiae (Ephydatia muelleri, Carteriospongia foliascens) and Hexactinellida (Oopsacas minuta, Heterochone calyx), the latter two depicted in our Fig. 1 as a clade named Silicea based on the shared presence of spicules made of hydrated silica. These analyses place Porifera as sister group to all other metazoans, but bootstrap support is only 62% and relationships between the five groups of metazoans (Fig. 3d) are highly sensitive to outgroup selection. In contrast to trees based on nuclear housekeeping genes (Sperling et al. 2009a, 2010) that depict sponges as a paraphyletic group, the monophyly of Porifera is strongly supported in all the analyses by Philippe et al. (2009). Importantly, while Placozoa is a monospecific phylum (but clearly composed of additional cryptic species; e.g. Voigt et al. 2004) and Ctenophora is well known to have a very long stem for the genes studied (Podar et al. 2001), the Philippe et al. (2009) study confirmed that the stems of Cnidaria and Porifera, as well as the two internal branches of the five-taxon tree, are extremely short (whereas the Bilateria stem is of moderate length). This mixture of branch lengths could indicate why the topology and rooting of the five-taxon tree are currently so unstable.
The first sponge genome was recently published (Srivastava et al. 2010), and the study included several phylogenomic analyses. In analyses limited to taxa for which complete genomes are available (lacking ctenophores because no ctenophore genome sequence has yet been published), the sponge was found to be sister group to the other sampled animals. Cnidarians were placed as sister group to Bilateria, and Placozoa as sister to that grouping. Analyses that included EST data for additional taxa, including more ctenophores, recovered Ctenophora as sister group to all other animals (Srivastava et al. 2010, supplementary information section S7), though the authors stressed the importance of additional taxon sampling, particularly among the outgroup taxa. Preliminary analyses of a draft ctenophore genome (Ryan et al. 2010) supported a clade comprised of Placozoa, Bilateria and Cnidaria (resolved as in Fig. 3b), to the exclusion of Porifera and Ctenophora. That study, however, did not differentiate between the alternative placements of Porifera or Ctenophora as sister group to the remaining Metazoa.
Acoelomorpha and Xenoturbella are a clade
Hejnol et al. (2009) corroborated previous suggestions that the acoel and nemertodermatid flatworms form a clade, Acoelomorpha, that is sister group to all other bilaterian animals, the latter being united under the taxonomic name Nephrozoa in recognition of the shared presence of an excretory system (Jondelius et al. 2002; Ruiz-Trillo et al. 1999, 2002; Sempere et al. 2007). Nephrozoa itself encompasses two putative clades whose status and membership have been debated since the 19th century: Protostomia and Deuterostomia. The Hejnol et al. (2009) study found diminishing evidence for the placement of the enigmatic Xenoturbella within Deuterostomia (Bourlat et al. 2003, 2006, 2009; Perseke et al. 2007); instead, all analyses placed Xenoturbella with Acoelomorpha, with which it shares several morphological features (Lundin 1998, 2001; Nielsen 2010; Pedersen and Pedersen 1986, 1988; Raikova et al. 2000; Westblad 1949). This phylogenetic position of Xenoturbella with Acoelomorpha as sister group to Nephrozoa was not recovered with phylogenomic studies that instead placed Xenoturbella among deuterostomes (Philippe et al. 2007; Philippe et al. 2011), a relationship also supported by ciliary ultrastructure (Franzén and Afzelius 1987; Pardos 1988).
The phylogenetic position of acoelomorphs + Xenoturbella will continue to be debated in light of new data (Philippe et al. 2011), but the most recent phylogenomic analyses agree with the morphological view relating Xenoturbella to acoels and nemertodermatids (e.g. Lundin 1998; Nielsen 2010). The study of Philippe et al. (2011) is based on three different datasets—mitochondrial protein coding genes, phylogenomic analyses and microRNA content—which all provide only weak evidence for a position of Acoelomorpha + Xenoturbellida with deuterostomes rather than more basally among Metazoa. The microRNA content presented in Philippe et al. (2011) is incongruent to the microRNA content of nemertodermatid Meara stichopi published elsewhere (Wallberg 2009), which instead supports a position of acoelomorphs before the protostome-deuterostome split. A deuterostome affinity of acoels is at odds with most morphological evidence. The organization (or lack) of organ systems in acoelomorphs and Xenoturbella, such as gross body morphology, the blind gut, non-epithelial gonads, lack of an excretory and vascular system, a simple brain, and the lack of a dorsal or ventral centralized nerve chord, shows no similarities with deuterostomes (Hejnol and Martindale 2008a). Furthermore, acoelomorphs and Xenoturbella do not show any traces of deuterostome characters, such as gill slits, that one would expect to be present even in highly derived lineages (such as the chorda in ascidians). If a deuterostome position does become established for this clade then it would suggest that its members have undergone dramatic losses of features.
Molecular dating generally interprets the divergences between most bilaterian phyla to have taken place at least during the Ediacaran Period, 635–542.5 Mya (Peterson et al. 2008), or even earlier, in the Cryogenian (Blair 2009); see the section entitled “A bilaterian chronogram” below. Macroscopic fossils from the latter half of the Ediacaran (from ca. 575 Mya) have been assigned by palaeontologists to the stem- or crown-groups of Bilateria (Xiao and Laflamme 2009), and some have been placed in the stem groups of particular bilaterian subclades, such as Mollusca or Panarthropoda (Peterson et al. 2008). The phylogenetic affinities of the Ediacaran macrofossils remain subject to highly divergent opinions, and the case for Bilateria remains ambiguous from the body fossils alone (the trace fossil record for at least the terminal part of the Ediacaran, from 555 Mya, provides a stronger case for Bilateria; Jensen et al. 2005). It appears likely that Ediacaran macrofossils will continue to be slotted into phylogenies inferred from molecular data rather than fundamentally contributing to tree reconstruction themselves.
Deuterostomia
Deuterostomes are defined as a group of animals in which through development the blastopore typically becomes the anus in the adult, while the mouth develops as a new opening from the end of the archenteron, but the fate of the blastopore shows considerable intraphyletic variation (Nielsen 2001). The composition of Deuterostomia has also changed through time, with some of the lophophorate groups (especially Brachiopoda and Phoronida) still being considered deuterostomes by some authors due to the scarcity of opposing morphological arguments, although all molecular studies place them with the protostome phyla (see below). Some morphological arguments for protostome affinities were put forward by Hejnol (2010), such as a derived trochophore larva and similarities in chaetal structures. Likewise, putative deuterostome affinities of chaetognaths have lost strength after the emergence of phylogenomic analyses (Marlétaz et al. 2006; Matus et al. 2006; see additional discussion on Chaetognatha below). Currently, Echinodermata and Hemichordata are grouped as the clade Ambulacraria, principally based on phylogenomics, while the three chordate lineages, Cephalochordata, Urochordata and Craniata, also form a clade. Ambulacraria is also supported by several exclusive gene expression patterns not shared by Chordata (Brown et al. 2008), although recent data on enteropneust neurogenesis have been argued to instead support a sister-group relationship between Hemichordata and Chordata (Kaul and Stach 2010). Within Chordata, Olfactores groups Urochordata and Craniata (Delsuc et al. 2006), and in molecular datasets only the position of Cephalochordata has received minor instability, with a brief placement as sister group to Echinodermata (Delsuc et al. 2006) soon corrected in favour of sister-group relations to Olfactores by subsequent phylogenomic analyses (e.g. Delsuc et al. 2008; Dunn et al. 2008; Hejnol et al. 2009).
The discovery of putative deuterostome fossils from the Cambrian, such as vetulicolians (Aldridge et al. 2007; Shu et al. 2001), yunnanozoans (Mallatt and Chen 2003; Shu et al. 2003), vetulocystids (Shu et al. 2004), and cambroemids (Caron et al. 2010), has added to evidence provided by pre-radial stem group echinoderms (Smith 2005) that external gill slits are a primitive deuterostome character lost during the early evolution of echinoderms (see reviews by Shu et al. 2010; Smith and Swalla 2009). The deuterostome affinities of at least some of those groups (notably the vetulicolians and yunnanozoans) are disputed by other palaeontologists, who posit that possible moult assemblages indicate a relationship to Ecdysozoa (e.g. Bergström 2010).
Despite the long-standing molecular-based acceptance of the composition of Deuterostomia and monophyly of Ambulacraria (e.g. Winchell et al. 2002), some authors have challenged the monophyly of Chordata (e.g. Delsuc et al. 2006; Mallatt et al. 2010; Mallatt and Winchell 2007), and many analyses limited to few genes find non-monophyly of either protostomes or deuterostomes (e.g. Mallatt et al. 2010). Perhaps the newest challenge, and still unsettled, is the position of Xenoturbella, once postulated to have an affinity to hemichordates (Pedersen and Pedersen 1986, 1988; see discussion above).
Possibly the last deep relationship to have been settled within Deuterostomia is the monophyly of Hemichordata, a group that had been considered as paraphyletic by some authors (e.g. Nielsen 2001 separated Pterobranchia and Enteropneusta as two phyla; Ax 2001 proposed paraphyly of Hemichordata as well as of Pterobranchia). All recent molecular analyses exploring a broad diversity within the phylum agree on the monophyly of Hemichordata (e.g. Cannon et al. 2009; Holland et al. 2009).
The main protostome lineages and the position of Chaetognatha
Evidence for the monophyly of protostomes has typically come from developmental characters such as the fate of the blastopore and the mode of formation of the mesoderm (Nielsen 2001), though new character sources such as a suite of novel microRNA families (Wheeler et al. 2009) strengthen the case for Protostomia as a clade. Traditionally protostomes had been depicted as a paraphyletic assemblage of worm-like animals that share a dorsal (or circumesophageal) brain connected to a ventral, often paired longitudinal nerve cord. Given the lack of a ventral centralized nerve cord in acoels and nemertodermatids (e.g. Raikova et al. 2004a, b) and the current phylogenetic framework for metazoans, we follow Giribet et al. (2009) in excluding acoels and nemertodermatids from Protostomia in the following discussion. Two clades encompass most of the diversity within Protostomia: Ecdysozoa and Spiralia (Fig. 1).
A taxon that unites moulting protostomes, Ecdysozoa, is currently recognized as monophyletic in most analyses. Skepticism about its validity has particularly come from genome analyses with only a few taxa several of which have rejected Ecdysozoa in favour of a taxon that unites coelomate animals; while the corresponding ‘Coelomata hypothesis’ is a traditional one, little morphological evidence has been offered for its support in modern times. The Coelomata hypothesis relates arthropods more closely to vertebrates than to nematodes (Blair et al. 2002; Dopazo et al. 2004; Longhorn et al. 2007; Philip et al. 2005; Rogozin et al. 2007; Wolf et al. 2004). Numerous reanalyses of those data and, notably, more comprehensively sampled datasets, have in turn dismissed Coelomata as an artefact of poor taxon sampling (i.e. distant outgroups and systematic artefactual attraction of nematodes to phylogenetically distant taxa at the base of Bilateria) (Dunn et al. 2008; Hejnol et al. 2009; Holton and Pisani 2010; Irimia et al. 2007; Philippe et al. 2005).
The name Spiralia (see discussions on this name vs. Lophotrochozoa in Giribet et al. 2009 and Hejnol 2010) was first coined by Schleip (1929) because of the stereotypical spiral development that occurs only within this clade (Maslakova et al. 2004a; Nielsen 2001). Spiralia contains all animals with spiral development in addition to some others that do not show this special mode of development. This character, like many others within Metazoa, shows homoplasy at several levels, apparently in the form of secondary reduction (Hejnol 2010)—but never as convergence outside the clade. This indicates that any animal with spiral development is an unambiguous member of the clade Spiralia, whereas absence does not necessarily invalidate membership, as this type of development has been lost several times.
The position of the chaetognaths (‘arrow worms’) remains one of the major controversies in bilaterian phylogenetics. Among the range of alternative affinities proposed for this group over the long history of debate, recent studies based on both morphology (Harzsch and Müller 2007) and phylogenomics (Dunn et al. 2008; Hejnol et al. 2009; Marlétaz et al. 2006; Matus et al. 2006) have strongly converged on chaetognaths being early diverging members of Protostomia, even though the latter show what has often been recognized as ‘deuterostome-like’ development (but see Kapp 2000). Beyond this, however, there is little consensus as to their exact position. Depending on character and taxon sampling, they have been placed either as sister group to Spiralia or within the latter (Dunn et al. 2008, Helmkampf et al. 2008b; Matus et al. 2006;), within Ecdysozoa (Baguñà et al. 2008; Helmkampf et al. 2008a; Paps et al. 2009b), or as sister group to Spiralia + Ecdysozoa (Dunn et al. 2008; Marlétaz et al. 2006; Matus et al. 2006). The nervous system of chaetognaths has recently been found to be similar to that of other protostomes in having a typical circumoral arrangement of the anterior CNS (Harzsch and Müller 2007). Resolving the placement of Chaetognatha is critical to the reconstruction of some of the most basic developmental characters for bilaterians, including cleavage mode and the fate of the blastopore.
Ecdysozoans and the status of Panarthropoda and Cycloneuralia
The ‘Ecdysozoa vs. Articulata’ debate of the middle part of the past decade (e.g. Giribet 1999, 2003, 2004; Jenner and Scholtz 2005; Pilato et al. 2005; Schmidt-Rhaesa 2004, 2006, 2007; Schmidt-Rhaesa et al. 1998; Scholtz 2002, 2003; Zrzavý 2001) is no longer a debate from the perspective of molecular data. The monophyly of Ecdysozoa, together with the alliance of annelids with molluscs and other spiralians, rather than with arthropods (Eernisse et al. 1992) as predicted by the Articulata hypothesis, is supported by an ever-growing series of analyses that have used different genes and diverse combinations of data and varied analytical approaches (Telford et al. 2008). In contrast, Articulata is not supported by any kind of molecular evidence.
Although the idea of arthropods as being allied to nematodes has a long history (reviewed by Scholtz 2002), the union of Panarthropoda and Cycloneuralia as Ecdysozoa (Aguinaldo et al. 1997) was originally established using data from the small nuclear ribosomal subunit (18S rRNA) and has been strengthened by numerous subsequent analyses using larger taxonomic samples of nuclear ribosomal genes (e.g. Giribet and Ribera 1998; Giribet and Wheeler 1999; Mallatt and Giribet 2006; Mallatt et al. 2004, 2010; Petrov and Vladychenskaya 2005; Zrzavý et al. 1998). Likewise, Ecdysozoa is a clade when sequences for small and large nuclear ribosomal RNA are analyzed together with 11 nuclear protein-coding genes (Baguñà et al. 2008), or when they are combined with complete mitochondrial genomes and nuclear protein-coding genes (Bourlat et al. 2008). Other kinds of molecular data that support Ecdysozoa are myosin heavy chain II (Ruiz-Trillo et al. 2002), Na+/K+-ATPase subunit (Kusche et al. 2005), a novel microRNA family (Wheeler et al. 2009), nuclear housekeeping genes (Helmkampf et al. 2008a, b), Hox gene signatures (Balavoine et al. 2002; de Rosa et al. 1999), mitochondrial genomics (Rota-Stabelli et al. 2010b), a combined analysis of 71 protein-coding genes (Philippe et al. 2005), and several other recent phylogenomic studies (e.g. Dunn et al. 2008; Hejnol et al. 2009; Holton and Pisani 2010; Philippe et al. 2007; ribosomal protein analyses of Bleidorn et al. 2009).
In addition to the body of molecular support, ecdysozoans share a set of characters related to the cuticle and its moulting. Where documented, moulting in the ecdysozoan phyla is induced by similar ecdysteroids, and all members of the group lack locomotory cilia—they lack ciliation on any external surface. Although some authors have attempted to correlate the presence of a thick cuticle with the lack of locomotory cilia, the presence of both in gastrotrichs (e.g. Ruppert 1991) corroborates their validity as independent phylogenetic characters. The layering of the cuticle in ecdysozoans shows detailed similarities, including differentiated epi-, exo- and endocuticle, with the first of these itself being trilaminate (Schmidt-Rhaesa et al. 1998). All Ecdysozoa for which the data have been documented share a unique tissue-specific immunoreactive marker, horseradish peroxidase (HRP), in the neural tissue (Haase et al. 2001).
Two main subgroups of Ecdysozoa have been retrieved in many analyses: Cycloneuralia and Panarthropoda. Cycloneuralia (also referred to as Introverta), named for the collar-shaped circumesophageal brain shared by its members, unites Nematoida (a widely recognized clade composed of Nematoda + Nematomorpha) with Priapulida, Kinorhyncha and Loricifera, the latter three often being grouped as a putative clade named Scalidophora. Panarthropoda sensu Nielsen (1995)—also named Ailopoda by Hou and Bergström (2006)—groups Arthropoda with Onychophora and Tardigrada, implying a single origin of paired ventrolateral segmental appendages in tardigrades, onychophorans and arthropods. The name Arthropoda is variably applied to Onychophora + Euarthropoda. Herein, we follow the prevalent usage in English-language publications of restricting the name Arthropoda to the euarthropods.
A result of several phylogenomic analyses that has challenged the monophyly of both Cycloneuralia and Panarthropoda is an affinity between nematodes and tardigrades (Hausdorf et al. 2010; Hejnol et al. 2009; Meusemann et al. 2010; Philippe et al. 2007; Roeding et al. 2007, 2009). Although some similarities, e.g. the myo-epithelial cells in the pharynx bulb, have long been observed among nematodes, tardigrades and loriciferans (Kristensen 2003), the standard morphological position of tardigrades over the past decade or two has been in the context of panarthropod monophyly, with tardigrades resolved either as sister group to Onychophora + Arthropoda (anatomical and phylogenomic arguments reviewed by Edgecombe 2010) or as sister group to Arthropoda (according to the Tactopoda hypothesis; Budd 2001) with Onychophora the sister group of that assemblage. The choice between Tardigrada + Nematoda and Tardigrada + (Onychophora + Arthropoda) is sensitive to the model used in maximum likelihood analyses and to taxon sampling (Dunn et al. 2008; Rota-Stabelli et al. 2010a, b), and alternative relationships of tardigrades to either nematodes or onychophorans are likewise found when broad taxon sampling is used in parsimony-based direct optimization analyses (Park et al. 2006).
The tardigrade-nematode attraction is tied to a recurring problem of establishing the monophyly of Cycloneuralia with molecular datasets. A few analyses have retrieved a partial ecdysozoan clade that resolves priapulids with Arthropoda or Panarthropoda while placing Nematoda in a phylogenetically distant position (mitogenomic datasets of Bleidorn et al. 2009; Rota-Stabelli et al. 2010b; Webster et al. 2006, 2007). Existing phylogenomic samples that include members of most major ecdysozoan groups and agree on the monophyly of Ecdysozoa have resolved Cycloneuralia as strictly monophyletic (Dunn et al. 2008: Bayesian trees after unstable taxa were deleted), as monophyletic if tardigrades are accepted as an ingroup taxon (Roeding et al. 2009), or as paraphyletic with respect to Panarthropoda., e.g. Nematoda closer to panarthropods than to scalidophorans (Rota-Stabelli et al. 2010b). Accordingly, pending a stable resolution of the tardigrade-nematode question, our summary tree (Fig. 1) leaves the monophyly of Cycloneuralia and Panarthropoda open.
The Scalidophora concept was formulated based largely on shared details of the introvert in priapulids, kinorhynchs and loriciferans, and each of the three possible interrelationships among the three groups has been advocated in the recent literature based on morphology (reviewed by Neuhaus and Higgins 2002). However, the sparse molecular data for loriciferans ally the group with Nematomorpha rather than with Kinorhyncha and/or Priapulida, and imply that morphological characters previously considered as synapomorphic for the scalidophoran taxa would instead be conditions for Cycloneuralia (Sørensen et al. 2008; see Park et al. 2006 for a less precise hypothesis), if not even for Ecdysozoa as a whole. The sister-group relationship between Priapulida and Kinorhyncha has been found in multilocus analyses (e.g. Mallatt and Giribet 2006; Paps et al. 2009b) as well as in phylogenomic ones (Dunn et al. 2008; but see Hejnol et al. 2009), although these analyses did not include molecular sequence data from loriciferans, whose members have recently been found to constitute the first metazoans to live in permanently anoxic conditions (Danovaro et al. 2010).
The Cambrian fossil record is an additional source of data bearing on scalidophoran/cycloneuralian interrelationships. Large macrofaunal fossils similar to loriciferans have recently been documented from the Cambrian Sirius Passet Lagerstätte of Greenland (Peel 2010), supplementing the record of loricate larvae known from Cambrian phosphatic microfossils (Maas et al. 2009). Stem-group priapulids such as palaeoscolecids are known from detailed anatomical preservation including cuticular structure and sensilla (Harvey et al. 2010), and three-dimensionally preserved embryos allow inferences on life history strategies in putative stem-group scalidophorans (Dong 2007).
Spiralia
The first phylogenomic studies to include lophophorates indicated that Lophotrochozoa is nested within a clade of animals with spiral cleavage (Helmkampf et al. 2008a, b) or perhaps is equivalent to Spiralia (Dunn et al. 2008; Hejnol et al. 2009), the name adopted here (as in von Döhren and Bartolomaeus 2007; Giribet 2002, 2008; Giribet et al. 2009; Hausdorf et al. 2007; Helmkampf et al. 2008b; Henry et al. 2007; Witek et al. 2009).
Spiralia has been suggested to comprise two putative clades, Platyzoa (Cavalier Smith 1998) and Trochozoa (Roule 1891). The name Trochozoa is preferred (see Giribet et al. 2000; Rouse 1999) over the more recently coined Eutrochozoa (Ghiselin 1988) used by some authors (Eernisse et al. 1992; Valentine 2004). Whether or not Platyzoa is a clade (e.g. Dunn et al. 2008; Giribet et al. 2000; Glenner et al. 2004; Hausdorf et al. 2010; Todaro et al. 2006; Zrzavý 2003) remains unclear, as many analyses provide low support and/or unstable relationships for the exact position of the ‘platyzoan’ phyla, although most authors tend to group them in a clade or in a grade giving rise to Trochozoa.
The identification by Dunn et al. (2008) of a core set of stable taxa within Spiralia whose relationships are well supported provides a more detailed picture of Platyzoa. The only stable taxon (as measured by leaf stability metrics) putatively assigned to Platyzoa was Platyhelminthes (Dunn et al. 2008, Fig. 2), which was found to be the sister group of Trochozoa with strong support in analyses restricted to stable taxa. All other platyzoans, e.g. the members of Gnathifera (sampled as Gnathostomulida and Rotifera) and Gastrotricha, were unstable in these analyses (Dunn et al. 2008, Fig. 1 and supplement), and their position could not be resolved with confidence. The platyzoan taxa investigated to date have relatively long branches, which has led some authors to suspect that support for the group is a systematic error (e.g. Telford 2006). The well-supported position of Platyhelminthes as sister group to Trochozoa (which cannot be the result of long-branch attraction, since Trochozoa does not contain taxa with long branches) may serve as an anchor attracting long-branch taxa whose placement is not based on strong signals. The resolution of this problem (including tests of this specific hypothesis) will require a two-pronged strategy of greatly improved taxon sampling within putative platyzoan groups, and detailed investigations into their positions designed specifically to identify systematic error. This part of the metazoan tree is seen as one of the most exciting areas of animal phylogenetic research, and as one that has been difficult to approach from the genomics point of view because of the small size and difficulties of working with many of its members.
Even if Platyzoa were a well-supported clade, an unequivocal morphological apomorphy is hard to delineate. The putative clade contains a series of acoelomate or pseudocoelomate animals—no coelomates belong to this group—some with special types of jaws formed of cuticularized rods, this group being named Gnathifera (Gnathostomulida, Micrognathozoa, and Rotifera) (Ahlrichs 1995; Kristensen and Funch 2000; Sørensen 2003). Some phylogenomic support for the monophyly of Gnathifera is emerging (Witek et al. 2009), albeit with indecisive resolution as to their placement relative to other Platyzoa. Platyzoa also includes other types of flat worms, including Platyhelminthes and Gastrotricha. Gastrotrichs have previously been suggested to be a basal lineage within Ecdysozoa or Cycloneuralia (Nielsen 2001; Schmidt-Rhaesa et al. 1998; Zrzavý 2003), based on several ecdysozoan traits including the cuticle and a circumpharyngeal brain. However, the neuropil of the circumpharyngeal brain of cycloneuralians maintains its size all around the pharynx (Schmidt-Rhaesa 2007), whereas the gastrotrich brain is instead composed of a ventral plus one or two dorsal commissures (Hochberg and Atherton 2011; Rothe and Schmidt-Rhaesa 2009). Thus, although both kinds of brains are wrapped around the pharynx, their homology is questionable. Similarities truly exist between the gastrotrich and the cycloneuralian (in particular the nematode) pharynx, which in both cases is muscular, triradial and highly cuticularized. On the other hand such pharynxes are also found elsewhere within the Metazoa, and even within the Platyzoa among some rotifers (de Beauchamp 1965), and they might have evolved convergently on several occasions. It is difficult to point out any clear-cut platyzoan autapomorphies, but all members of the group, with the exception of polyclad flatworms and the parasitic acanthocephalan rotifers, are strictly direct developers.
Trochozoa contains those groups with a typical trochophore larva, defined as having a ciliary band known as a prototroch (Rouse 1999), as occurs in Annelida sensu lato and Mollusca. The larva of a nemertean has been interpreted as a modified trochophore with a vestigial prototroch (Maslakova et al. 2004b), which further supports the inclusion of Nemertea in Trochozoa. That said, many Trochozoa do not develop through a trochophore stage (e.g. Brachiopoda, Phoronida, and most nemerteans). Even within groups in which a trochophore is widespread (such as Annelida and Mollusca) there are clear cases of it being lost (e.g. clitellate annelids and cephalopod molluscs). Trochophores are thought to occur outside Trochozoa, as delineated in Fig. 1 in only one case, Entoprocta (see Nielsen 2001), which has been suggested to be the sister group of Mollusca (Haszprunar 1996, 2000; Haszprunar and Wanninger 2008; Wanninger 2008), based especially on shared features of the foot sole of the larva of Loxosomella murmanica and Neomeniomorpha, but also on the presence of chitinous non-moulted cuticle, the sinus circulatory system and a number of neural features (e.g. Haszprunar and Wanninger 2008). Considering the contradicting molecular results, further work on the placement of the group and the cell lineage of its ‘prototroch’ is needed.
From the time of their original description (Funch and Kristensen 1995), affinities of Cycliophora with entoprocts and bryozoans have been identified as highly likely (also see Giribet et al. 2004). A sister-group relationship between entoprocts and cycliophorans was strongly supported in the first phylogenomic analysis to include Cycliophora (Hejnol et al. 2009). This clade was placed as the sister group to Bryozoa (see Polyzoa in Fig. 2 of Hejnol et al. 2009), although with low support. One of the cycliophoran larval forms, the chordoid larva, has been interpreted as a modified trochophore (Funch 1996), but its homology with true trochophores, such as that of Entoprocta, needs further study. The overall neuroanatomical condition of the cyliophoran chordoid larva much more closely resembles the situation of adult rather than larval stages of a number of spiralian taxa (Neves et al. 2010).
Annelids represent perhaps one of the most interesting and unstable areas of bilaterian phylogeny due to the large number of species and disparate body plans that have been assigned to this phylum through history. While certain apomorphies of the phylum are well established (Rouse and Pleijel 2007), its members are capable of modifying each and every single one of them. For example, there are annelids without segments (e.g. Echiura), without chaetae (e.g. Dinophilidae, Hirudinea) or without nuchal organs (e.g. Clitellata); at the same time, segments of similar identity are present in other phyla (e.g. Arthropoda and Chordata; see Seaver 2003), and ultrastructurally identical chaetae are present in brachiopods (Lüter 2000). Therefore, the composition of Annelida has been in flux, and several taxa once treated as separate phyla are now considered as parts of Annelida, including Pogonophora, Vestimentifera, Echiura, and—more contentiously as to whether it is sister to Annelida or a part of the annelid ingroup—Sipuncula (e.g. Dordel et al. 2010; Dunn et al. 2008; Hejnol et al. 2009; Mwinyi et al. 2009; Sperling et al. 2009b; Struck et al. 2007). Other taxa traditionally treated as annelids, such as Diurodrilus, are now considered as of uncertain kinship (Worsaae and Rouse 2008), and whether Myzostomida are related to annelids or to platyzoans is still in flux (Bleidorn et al. 2007, 2009; Eeckhaut et al. 2000; Hejnol et al. 2009; Mallatt et al. 2010; Zrzavý et al. 2001). For other problematica with suggested annelid affinity, such as Jennaria (Rieger 1991), Lobatocerebrum (Rieger 1980) and the parasitic Orthonectida (Halanych 2004; Slyusarev and Kristensen 2003), the lack of informative morphological and especially molecular data may explain why these taxa are generally not even considered in phylogenetic analyses.
Do phylogenomic trees conflict with morphology?
Our review has emphasized recent advances in metazoan phylogeny contributed mostly by molecular systematics, and in particular by phylogenomic approaches. Figure 1 is a summary of clades that have been recovered repeatedly in well-sampled phylogenomic analyses. We do not, however, make a distinction between this topology and ‘a’ morphological cladogram for Metazoa, because the groups in Fig. 1 are in large part the ones recovered by morphologists. Well-established ‘phylogenomic’ clades such as Bilateria, Protostomia, Deuterostomia, Spiralia, and Trochozoa were identified, of course, by 19th century zoologists based on anatomical and developmental evidence. The composition of Protostomia and Deuterostomia varies among authors with respect to the position of the ‘lophophorates’, but the corresponding disagreement is individual rather than between morphologists and molecular biologists per se, as many morphologists and paleontologists have suggested relationships of brachiopods to molluscs, or of bryozoans to entoprocts and cycliophorans, therefore implicitly placing ‘lophophorates’ in Protostomia. In most instances of phylogenomic topologies that have been truly novel and indicative of interpretations of anatomical character evolution very different from what had been anticipated by morphologists, such as the resolution of Ctenophora as the sister group of all other metazoans (Dunn et al. 2008), the ‘surprising’ result has been contradicted in other phylogenomic studies in favour of a more traditional topology (e.g. Philippe et al. 2009 for Porifera as sister group of Eumetazoa). Figure 1 includes a few clades that have certainly contradicted morphological orthodoxy, such as the identification of Olfactores in favour of a cephalochordate + craniate clade, or Ambulacraria in favour of Hemichordata as sister group of Chordata. That said, the ‘phylogenomic’ groupings are ones identified and named based on morphological arguments, in the case of Ambulacraria well over a century ago, and Olfactores emerged from a palaeontological hypothesis. Ecdysozoa is similar in many respects. It is indisputable that Ecdysozoa runs against the grain of two centuries of morphology-based classification, the era of nearly universal endorsement of Articulata (though with dissent from those who recognized characters that speak to a closer relationship between nematodes and arthropods; see Scholtz 2002 for historical precursors of the Ecdysozoa concept). However, it is a distortion to view Ecdysozoa as a ‘molecules versus morphology’ argument, because Ecdysozoa receives solid anatomical support from characters of the cuticle and its moulting. Perhaps the most unanticipated grouping included in Fig. 1, from the perspective of morphology, is Platyzoa. As discussed in the section on Spiralia above, unambiguous morphological or developmental apomorphies for a platyzoan clade are unknown and the potentially artefactual attraction of its members in molecular analyses must receive further testing.
We consider it a fair summary of the state of play to say that that the conflict between different phylogenomic datasets and between different morphological datasets with respect to metazoan relationships is not materially different from the respectively conflict (or, conversely, congruence) with each other.
A bilaterian chronogram: fossil-calibrated divergence times
Molecular dating with paleontological calibration provides a basis for estimating the divergence times of the major lineages of Bilateria (for recent examples and citations of earlier works, see Blair 2009; Peterson et al. 2008). Figure 4 is a chronogram based on the most likely topology in the 64-taxon Bayesian analysis of Dunn et al. (2008) using relaxed-clock methods for a 150-gene sample. Because of the substantial incongruence among the relationships between the main non-bilaterian lineages in phylogenomic studies to date (Fig. 1), the topology used for dating is for Bilateria alone.

Calibration to constraints was performed by penalized likelihood (Sanderson 2002), as implemented in r8s v1.71 (Sanderson 2003), with a truncated Newton (TN) algorithm. The optimization of the smoothing parameter was obtained using the cross-validation feature in r8s following the instructions of the program manual (available at http://loco.biosci.arizona.edu/r8s/). All analyses passed the gradient check implemented in r8s. Calibration points were assigned from minimum dates for branch points in the tree based on the oldest well-corroborated fossil(s) for clades with reliable palaeontological data (Table 1; Electronic supplementary material). A maximum of 635 Mya was set for Bilateria. This date was used by Peterson et al (2008) as a maximum for Eumetazoa based on palaeoecological changes associated with Marinoan deglaciation. Because subsequently published studies have provided strong evidence for crown-group sponges by 635 Mya (demosponge biomarker data of Love et al. 2009), we have not imposed an Ediacaran (<635 Mya) maximum for all eumetazoans and restrict this constraint to Bilateria only.
The resulting chronogram is consistent with Ediacaran origins for many animal phyla, and with dates of 598, 603 and 618 Mya for Ecdysozoa, Spiralia and Protostomia, respectively (Fig. 4). We emphasise that the imposition of a maximum on bilaterian origins at the Cryogenian-Ediacaran transition provides a shorter “fuse” (Conway Morris 2000) to the bilaterian radiation than has been retrieved in other clock trees that either excluded maxima or set them farther back in time (e.g. Blair 2009). Under those conditions, divergences of the major bilaterian lineages are typically dated to the Cryogenian, forcing the fossil record to be interpreted as a poor chronicle of divergence times (many lineages that have no fossil record until ca. 520 Mya, in the early Cambrian, are predicted to have originated at least 125 My earlier than their first palaeontological appearances). Our analysis attempts to calibrate with more minima (divergences conservatively constrained by reliable fossil assignments) and few maxima. The chronogram converges on previous studies in predicting that such bilaterian groups as Arthropoda, Mollusca, and Chordata had stem groups extending to various depths in time in the Ediacaran.
The critical role of developmental biology for informing and testing systematic hypotheses
Developmental biology has provided insight into the interpretation of morphological and anatomical characters. The description of the development of an organism can provide a more detailed picture of a morphological character and can give insights into the homology of structures and their evolutionary origin. Recent advances have been mainly in the fields of cell lineage and fate mapping studies and in the area of molecular developmental biology. Modern studies of cell lineages during early embryogenesis, both using intracellular dye injections and 4D microscopy techniques, have provided detailed understanding of the development of several animals and have aided in providing sound hypotheses of homology. Detailed fate mapping studies of spiralian embryos have helped to unravel the evolutionary origin and modifications of larval ciliary bands, a character often used in phylogenetic reconstructions. Such studies have allowed tracing the evolutionary history of the prototroch, including its loss in some nemertean larva (Maslakova et al. 2004a, b) and its modification to a feeding structure in the veliger larva of gastropods (Hejnol et al. 2007). High-resolution fate maps deliver a complex data-set for comparisons between major animal clades (see Hejnol et al. 2007; Meyer et al. 2010) and can also show the variation of developmental patterns, e.g. cell fate changes of the mesentoblast (4d cell) (Meyer et al. 2010). Small, non-injectable embryos can be studied by 3D timelapse microscopy (4D microscopy) (Hejnol and Schnabel 2005, 2006) and thus allow comparisons of fate maps across all Metazoa.
Another level for comparisons of structures between species has been added by studies of important transcription factors and signalling molecules that trigger the development of these structures. Answers to old questions such as the origin of the mesodermal germ layer may be indicated by the finding that mesodermal genes are expressed in the endoderm of diploblastic animals (Martindale et al. 2004). Specific transcription factors can help to determine the homology of structures, such as the mouth in cnidarians, acoelomorphs, protostomes and deuterostomes (Arendt et al. 2001; Hejnol and Martindale 2008b). Similarly, it has helped to understand the evolution of the bilaterian anus, possibly from a gonopore (Hejnol and Martindale 2008b). Not only organ systems and larger structures can be traced back to their evolutionary origin using gene expression studies, but also single cell types, e.g. photoreceptors, formerly described exclusively by ultrastructural studies, are now characterized by the expression of specific opsins that correlate with the cell morphology (Arendt et al. 2004; Passamaneck et al. 2011).
However, there are pitfalls to be avoided when interpreting gene expression patterns. Not every complex structure can be homologized solely by gene expression patterns. It has been shown that ‘simple’ animals such as the sea anemone Nematostella vectensis have the complete set of bilaterian WNT genes (Kusserow et al. 2005) and that the complex segmented arthropod legs follow the same patterning system as the non-articulated legs of onychophorans (Janssen et al. 2010) which was co-opted from a head-axis patterning system (Lemmons et al. 2010) in the arthropod lineage. To get a better understanding of the developmental changes during evolution, more studies of ‘minor’ taxa are needed. Especially by looking at simple structured organisms will we obtain a better understanding of the branches of the phylogenetic tree on which important gains and losses have taken place.
Conclusions
It has been almost 150 years since Ernst Haeckel proposed his first animal tree of life. Since then, developments in anatomical study, homology and taxon definition, phylogenetic methods and numerical analysis of explicit data-sets have contributed towards a robust animal phylogeny that is converging towards a single tree from what seemed to be a forest of possible relationships. Work, and not a negligible amount, remains to be done to settle important debates, such as concerning the relationships at the base of the animal tree, and those of more derived taxa with otherwise unstable positions (e.g. chaetognaths, platyzoans), but many clades appear well established at last. Resolution within protostomes is now clear between Spiralia (itself divided in Trochozoa and the possible clades Polyzoa and Platyzoa—or a grade of flat worms leading to Trochozoa) and Ecdysozoa. Deuterostome composition is restricted to five phyla that group as two main lineages, Ambulacraria and the three chordate phyla, with the only remaining issue being the position of Xenoturbellida. There is no doubt that metazoan systematists will continue refining the position of many of the branches and twigs in this entangled tree of animal life, but we are getting to a point where the main branches are becoming well understood, the prerequisite to exploring other evolutionary questions in animal biology.
References
Adoutte, A., Balavoine, G., Lartillot, N., Lespinet, O., Prud’homme, B., & de Rosa, R. (2000). The new animal phylogeny: reliability and implications. Proceedings of the National Academy of Sciences of the USA, 97, 4453–4456.
Aguinaldo, A. M. A., Turbeville, J. M., Lindford, L. S., Rivera, M. C., Garey, J. R., Raff, R. A., et al. (1997). Evidence for a clade of nematodes, arthropods and other moulting animals. Nature, 387, 489–493.
Ahlrichs, W. H. (1995). Seison annulatus und Seison nebaliae—Ultrastruktur und Phylogenie. Verhandlungen der Deutschen Zoologischen Gesellschaft, 88, 155.
Aldridge, R. J., Hou, X.-G., Siveter, D. J., Siveter, D. J., & Gabbott, S. E. (2007). The systematics and phylogenetic relationships of vetulicolians. Palaeontology, 50, 131–168.
Arendt, D., Technau, U., & Wittbrodt, J. (2001). Evolution of the bilaterian larval foregut. Nature, 409, 81–85.
Arendt, D., Tessmar-Raible, K., Snyman, H., Dorresteijn, A. W., & Wittbrodt, J. (2004). Ciliary photoreceptors with a vertebrate-type opsin in an invertebrate brain. Science, 306, 869–871.
Ax, P. (1996). Multicellular animals. A new approach to the phylogenetic order in nature. Volume I. Berlin: Springer.
Ax, P. (2001). Das System der Metazoa III. Ein Lehrbuch der phylogenetischen Systematik. Stuttgart: Spektrum Akademischer Verlag.
Backeljau, T., Winnepenninckx, B., & De Bruyn, L. (1993). Cladistic analysis of metazoan relationships: a reappraisal. Cladistics, 9, 167–181.
Baguñà, J., Martinez, P., Paps, J., & Riutort, M. (2008). Back in time: a new systematic proposal for the Bilateria. Proceedings of the Royal Society / B, 363, 1481–1491.
Balavoine, G., de Rosa, R., & Adoutte, A. (2002). Hox clusters and bilaterian phylogeny. Molecular Phylogenetics and Evolution, 24, 266–373.
de Beauchamp, P. (1965). Classe des Rotifères. In P. P. Grassé (Ed.), Traité de Zoologie IV, 3 (pp. 1225–1379). Paris: Masson.
Bergström, J. (2010). The earliest arthropods and other animals. In M.-Y. Long (Ed.), Darwin’s heritage today. Proceedings of the Darwin 200 International Conference (pp. 29–42). Beijing: Higher Education Press.
Blair, J. E. (2009). Animals (Metazoa). In S. B. Hedges & S. Kumar (Eds.), The timetree of life (pp. 223–230). Oxford: Oxford University Press.
Blair, J. E., Ikeo, K., Gojobori, T., & Hedges, S. B. (2002). The evolutionary position of nematodes. BMC Evolutionary Biology, 2, 1–7.
Bleidorn, C., Eeckhaut, I., Podsiadlowski, L., Schult, N., McHugh, D., Halanych, K. M., et al. (2007). Mitochondrial genome and nuclear sequence data support Myzostomida as part of the annelid radiation. Molecular Biology and Evolution, 24, 1690–1701.
Bleidorn, C., Podsiadlowski, L., Zhong, M., Eeckhaut, I., Hartmann, S., Halanych, K. M., et al. (2009). On the phylogenetic position of Myzostomida: can 77 genes get it wrong? BMC Evolutionary Biology, 9, 150.
Bourlat, S. J., Nielsen, C., Lockyer, A. E., Littlewood, D. T., & Telford, M. J. (2003). Xenoturbella is a deuterostome that eats molluscs. Nature, 424, 925–928.
Bourlat, S. J., Juliusdottir, T., Lowe, C. J., Freeman, R., Aronowicz, J., Kirschner, M., et al. (2006). Deuterostome phylogeny reveals monophyletic chordates and the new phylum Xenoturbellida. Nature, 444, 85–88.
Bourlat, S. J., Nielsen, C., Economou, A. D., & Telford, M. J. (2008). Testing the new animal phylogeny: a phylum level molecular analysis of the animal kingdom. Molecular Phylogenetics and Evolution, 49, 23–31.
Bourlat, S. J., Rota-Stabelli, O., Lanfear, R., & Telford, M. J. (2009). The mitochondrial genome structure of Xenoturbella bocki (phylum Xenoturbellida) is ancestral within the deuterostomes. BMC Evolutionary Biology, 9, 107.
Brown, F. D., Prendergast, A., & Swalla, B. J. (2008). Man is but a worm: chordate origins. Genesis, 46, 605–613.
Budd, G. E. (2001). Tardigrades as ‘stem-group arthropods’: the evidence from the Cambrian fauna. Zoologischer Anzeiger, 240, 265–279.
Cannon, J. T., Rychel, A. L., Eccleston, H., Halanych, K. M., & Swalla, B. J. (2009). Molecular phylogeny of Hemichordata, with updated status of deep-sea enteropneusts. Molecular Phylogenetics and Evolution, 52, 17–24.
Caron, J.-B., Conway Morris, S., & Shu, D. (2010). Tentaculate fossils from the Cambrian of Canada (British Columbia) and China (Yunnan) reinterpreted as primitive deuterostomes. PLoS ONE, 5, e9586.
Carranza, S., Baguñà, J., & Riutort, M. (1997). Are Platyhelminthes a monophyletic group? An assessment using 18S rDNA sequences. Molecular Biology and Evolution, 14, 485–497.
Cavalier Smith, T. (1998). A revised six-kingdom system of life. Biological Reviews, 73, 203–266.
Conway Morris, S. (2000). The Cambrian “explosion”: slow-fuse or megatonnage? Proceedings of the National Academy of Sciences of the USA, 97, 4426–4429.
Copley, R. R., Aloy, P., Russell, R. B., & Telford, M. J. (2004). Systematic searches for synapomorphies in the model metazoan genomes give some support for Ecdysozoa after accounting for the idiosyncrasies of Caenorhabditis elegans. Evolution & Development, 6, 164–169.
Danovaro, R., Dell’Anno, A., Pusceddu, A., Gambi, C., Heiner, I., & Kristensen, R. M. (2010). The first Metazoa living in permanently anoxic conditions. BMC Biology, 8, 30.
Dellaporta, S. L., Xu, A., Sagasser, S., Jakob, W., Moreno, M. A., Buss, L. W., et al. (2006). Mitochondrial genome of Trichoplax adhaerens supports Placozoa as the basal lower metazoan phylum. Proceedings of the National Academy of Sciences of the USA, 103, 8751–8756.
Delsuc, F., Brinkmann, H., & Philippe, H. (2005). Phylogenomics and the reconstruction of the tree of life. Nature Reviews / Genetics, 6, 361–375.
Delsuc, F., Brinkmann, H., Chourrout, D., & Philippe, H. (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature, 439, 965–668.
Delsuc, F., Tsagkogeorga, G., Lartillot, N., & Philippe, H. (2008). Additional molecular support for the new chordate phylogeny. Genesis, 46, 592–604.
von Döhren, J., & Bartolomaeus, T. (2007). Ultrastructure and development of the rhabdomeric eyes in Lineus viridis (Heteronemertea, Nemertea). Zoology (Jena), 110, 430–438.
Dong, X. (2007). Developmental sequence of Cambrian embryo Markuelia. Chinese Science Bulletin, 52, 929–935.
Dopazo, H., & Dopazo, J. (2005). Genome-scale evidence of the nematode-arthropod clade. Genome Biology, 6, R41.
Dopazo, H., Santoyo, J., & Dopazo, J. (2004). Phylogenomics and the number of characters required for obtaining an accurate phylogeny of eukaryote model species. Bioinformatics, 20(Supplement 1), I116–I121.
Dordel, J., Fisse, F., Purschke, G., & Struck, T. H. (2010). Phylogenetic position of Sipuncula derived from multi-gene and phylogenomic data and its implication for the evolution of segmentation. Journal of Zoological Systematics and Evolutionary Research, 48, 197–207.
Dunn, C. W., Hejnol, A., Matus, D. Q., Pang, K., Browne, W. E., Smith, S. A., et al. (2008). Broad phylogenomic sampling improves resolution of the animal tree of life. Nature, 452, 745–749.
Edgecombe, G. D. (2010). Arthropod phylogeny: an overview from the perspectives of morphology, molecular data and the fossil record. Arthropod Structure & Development, 39, 74–87.
Eeckhaut, I., McHugh, D., Mardulyn, P., Tiedemann, R., Monteyne, D., Jangoux, M., et al. (2000). Myzostomida: a link between trochozoans and flatworms? Proceedings of the Royal Society of London, Series B, 267, 1383–1392.
Eernisse, D. J., Albert, J. S., & Anderson, F. E. (1992). Annelida and Arthropoda are not sister taxa: a phylogenetic analysis of spiralian metazoan morphology. Systematic Biology, 41, 305–330.
Egger, B., Steinke, D., Tarui, H., De Mulder, K., Arendt, D., Borgonie, G., et al. (2009). To be or not to be a flatworm: the acoel controversy. PLoS ONE, 4, e5502.
Farris, J. S. (1970). Methods for computing Wagner trees. Systematic Zoology, 19, 83–92.
Farris, J. S., Kluge, A. G., & Eckhardt, M. J. (1970). A numerical approach to phylogenetic systematics. Systematic Zoology, 19, 172–189.
Felsenstein, J. (1973). Maximum-likelihood estimation of evolutionary trees from continuous characters. American Journal of Human Genetics, 25, 471–492.
Field, K. G., Olsen, G. J., Lane, D. J., Giovannoni, S. J., Ghiselin, M. T., Raff, E. C., et al. (1988). Molecular phylogeny of the animal kingdom. Science, 239, 748–753.
Franzén, Å., & Afzelius, B. A. (1987). The ciliated epidermis of Xenoturbella bocki (Platyhelminthes, Xenoturbellida) with some phylogenetic considerations. Zoologica Scripta, 16, 9–17.
Funch, P. (1996). The chordoid larva of Symbion pandora (Cycliophora) is a modified trochophore. Journal of Morphology, 230, 231–263.
Funch, P., & Kristensen, R. M. (1995). Cycliophora is a new phylum with affinities to Entoprocta and Ectoprocta. Nature, 378, 711–714.
Gamulin, V., Muller, I. M., & Muller, W. E. (2000). Sponge proteins are more similar to those of Homo sapiens than to Caenorhabditis elegans. Biological Journal of the Linnean Society, 71, 821–828.
Ghiselin, M. T. (1988). The origin of molluscs in the light of molecular evidence. Oxford Surveys in Evolutionary Biology, 5, 66–95.
Giribet, G. (1999). Ecdysozoa versus Articulata, dos hipótesis alternativas sobre la posición de los Artrópodos en el reino Animal. In A. Melic, J. J. de Haro, M. Méndez, & I. Ribera (Eds.), Evolución y filogenia de Arthropoda (pp. 145–160). Zaragoza: Sociedad Entomológica Aragonesa.
Giribet, G. (2002). Current advances in the phylogenetic reconstruction of metazoan evolution. A new paradigm for the Cambrian explosion? Molecular Phylogenetics and Evolution, 24, 345–357.
Giribet, G. (2003). Molecules, development and fossils in the study of metazoan evolution; Articulata versus Ecdysozoa revisited. Zoology, 106, 303–326.
Giribet, G. (2004). ¿Articulata o Ecdysozoa?: una revisión crítica sobre la posición de los artrópodos en el reino animal. In J. E. Llorente Bousquets, J. J. Morrone, O. Yáñez Ordóñez, & I. Vargas Fernández (Eds.), Biodiversidad, taxonomía y biogeografía de artrópodos de México: Hacia una síntesis de su conocimiento. Volúmen IV (pp. 45–62). Mexico: UNAM, Facultad de Ciencias.
Giribet, G. (2008). Assembling the lophotrochozoan (=spiralian) tree of life. Philosophical Transactions of the Royal Society, Series B, 363, 1513–1522.
Giribet, G., & Ribera, C. (1998). The position of arthropods in the animal kingdom: a search for a reliable outgroup for internal arthropod phylogeny. Molecular Phylogenetics and Evolution, 9, 481–488.
Giribet, G., & Wheeler, W. C. (1999). The position of arthropods in the animal kingdom: Ecdysozoa, islands, trees, and the “parsimony ratchet”. Molecular Phylogenetics and Evolution, 13, 619–623.
Giribet, G., Distel, D. L., Polz, M., Sterrer, W., & Wheeler, W. C. (2000). Triploblastic relationships with emphasis on the acoelomates and the position of Gnathostomulida, Cycliophora, Plathelminthes, and Chaetognatha: a combined approach using 18S rDNA sequences and morphology. Systematic Biology, 49, 539–562.
Giribet, G., Sørensen, M. V., Funch, P., Kristensen, R. M., & Sterrer, W. (2004). Investigations into the phylogenetic position of Micrognathozoa using four molecular loci. Cladistics, 20, 1–13.
Giribet, G., Dunn, C. W., Edgecombe, G. D., & Rouse, G. W. (2007). A modern look at the animal tree of life. Zootaxa, 1668, 61–79.
Giribet, G., Dunn, C. W., Edgecombe, G. D., Hejnol, A., Martindale, M. Q., & Rouse, G. W. (2009). Assembling the spiralian tree of life. In M. J. Telford & D. T. J. Littlewood (Eds.), Animal evolution: Genes, genomes, fossils and trees (pp. 53–64). Oxford: Oxford University Press.
Glenner, H., Hansen, A. J., Sørensen, M. V., Ronquist, F., Huelsenbeck, J. P., & Willerslev, E. (2004). Bayesian inference of metazoan phylogeny: a combined molecular and morphological approach. Current Biology, 14, 1644–1649.
Haase, A., Stern, M., Wächtler, K., & Bicker, G. (2001). A tissue-specific marker of Ecdysozoa. Development Genes and Evolution, 211, 428–433.
Haeckel, E. (1866). Generelle Morphologie der Organismen. Berlin: Georg Reimer.
Halanych, K. M. (2004). The new view of animal phylogeny. Annual Reviews of Ecology, Evolution and Systematics, 35, 229–256.
Halanych, K. M., Bacheller, J. M., Aguinaldo, A. M. A., Liva, S. M., Hillis, D. M., & Lake, J. A. (1995). Evidence from 18S ribosomal DNA that lophophorates are protostome animals. Science, 267, 1641–1643.
Harvey, T. H. P., Dong, X., & Donoghue, P. C. J. (2010). Are palaeoscolecids ancestral ecdysozoans? Evolution & Development, 12, 177–200.
Harzsch, S., & Müller, C. H. G. (2007). A new look at the ventral nerve centre of Sagitta: implications for the phylogenetic position of Chaetognatha (arrow worms) and the evolution of the bilaterian nervous system. Frontiers in Zoology, 4, 14.
Haszprunar, G. (1996). The Mollusca: coelomate turbellarians or mesenchymate annelids? In J. D. Taylor (Ed.), Origin and evolutionary radiation of the Mollusca (pp. 1–28). Oxford: Oxford University Press.
Haszprunar, G. (2000). Is the Aplacophora monophyletic? A cladistic point of view. American Malacological Bulletin, 15, 115–130.
Haszprunar, G., & Wanninger, A. (2008). On the fine structure of the creeping larva of Loxosomella murmanica: additional evidence for a clade of Kamptozoa (Entoprocta) and Mollusca. Acta Zoologica, 89, 137–148.
Hausdorf, B., Helmkampf, M., Meyer, A., Witek, A., Herlyn, H., Bruchhaus, I., et al. (2007). Spiralian phylogenomics supports the resurrection of Bryozoa comprising Ectoprocta and Entoprocta. Molecular Biology and Evolution, 24, 2723–2729.
Hausdorf, B., Helmkampf, M., Nesnidal, M. P., & Bruchhaus, I. (2010). Phylogenetic relationships within the lophophorate lineages (Ectoprocta, Brachiopoda and Phoronida). Molecular Phylogenetics and Evolution, 55, 121–1127.
Hejnol, A. (2010). A twist in time—the evolution of spiral cleavage in the light of animal phylogeny. Integrative and Comparative Biology, 50, 695–706.
Hejnol, A., & Schnabel, R. (2005). The eutardigrade Thulinia stephaniae has an indeterminate development and the potential to regulate early blastomere ablations. Development, 132, 1349–1361.
Hejnol, A., & Schnabel, R. (2006). What a couple of dimensions can do for you: comparative developmental studies using 4D-microscopy—examples from tardigrade development. Integrative and Comparative Biology, 46, 151–161.
Hejnol, A., & Martindale, M. Q. (2008a). Acoel development supports a simple planula-like urbilaterian. Philosophical Transactions of the Royal Society, Series B, 363, 1493–1501.
Hejnol, A., & Martindale, M. Q. (2008b). Acoel development indicates the independent evolution of the bilaterian mouth and anus. Nature, 456, 382–386.
Hejnol, A., Martindale, M. Q., & Henry, J. Q. (2007). High-resolution fate map of the snail Crepidula fornicata: the origins of ciliary bands, nervous system, and muscular elements. Developmental Biology, 305, 63–76.
Hejnol, A., Obst, M., Stamatakis, A., Ott, M., Rouse, G. W., Edgecombe, G. D., et al. (2009). Assessing the root of bilaterian animals with scalable phylogenomic methods. Proceedings of the Royal Society, Series B, 276, 4261–4270.
Helmkampf, M., Bruchhaus, I., & Hausdorf, B. (2008a). Multigene analysis of lophophorate and chaetognath phylogenetic relationships. Molecular Phylogenetics and Evolution, 46, 206–214.
Helmkampf, M., Bruchhaus, I., & Hausdorf, B. (2008b). Phylogenomic analyses of lophophorates (brachiopods, phoronids and bryozoans) confirm the Lophotrochozoa concept. Proceedings of the Royal Society, Series B, 275, 1927–1933.
Hennig, W. (1950). Grundzüge einer Theorie der phylogenetischen Systematik. Berlin: Deutscher Zentralverlag.
Hennig, W. (1965). Phylogenetic systematics. Annual Review of Entomology, 10, 97–166.
Hennig, W. (1966). Phylogenetic systematics. Urbana: University of Illinois Press.
Henry, J. Q., Hejnol, A., Perry, K. J., & Martindale, M. Q. (2007). Homology of ciliary bands in spiralian trochophores. Integrative and Comparative Biology, 47, 865–871.
Hochberg, R., & Atherton, S. (2011). A new species of Lepidodasys (Gastrotricha, Macrodasyida) from Panama with a description of its peptidergic nervous system using, CLSM, anti-FMRFamide and anti-SCPB. Zoologischer Anzeiger. doi:10.1016/j.jcz.2010.12.002.
Holland, N. D., Jones, W. J., Ellena, J., Ruhl, H. A., & Smith, K. L. (2009). A new deep-sea species of epibenthic acorn worm (Hemichordata, Enteropneusta). Zoosystema, 31, 333–346.
Holton, T. A., & Pisani, D. (2010). Deep genomic-scale analyses of the Metazoa reject Coelomata: evidence from single- and multigene families analyzed under a supertree and supermatrix paradigm. Genome Biology and Evolution, 2, 310–324.
Hou, X., & Bergström, J. (2006). Dinocarids—anomalous arthropods or arthropod-like worms. In J. Rong, Z. Fang, Z. Zhou, R. Zhan, X. Wang, & X. Yuan (Eds.), Originations, radiations and biodiversity changes—Evidences from the Chinese fossil record (pp. 139–158, 847–850). Beijing: Science.
Irimia, M., Maeso, L., Penny, D., García-Fernandez, J., & Roy, S. W. (2007). Rare coding sequence changes are consistent with Ecdysozoa, not Coelomata. Molecular Biology and Evolution, 24, 604–1607.
Janssen, R., Eriksson, J. B., Budd, G. E., Akam, M., & Prpic, N.-M. (2010). Gene expression patterns in onychophorans reveal that regionalization predates limb segmentation in pan-arthropods. Evolution & Development, 12, 363–372.
Jenner, R. A. (2001). Bilaterian phylogeny and uncritical recycling of morphological data sets. Systematic Biology, 50, 730–742.
Jenner, R. A. (2004a). The scientific status of metazoan cladistics: why current research practice must change. Zoologica Scripta, 33, 293–310.
Jenner, R. A. (2004b). Towards a phylogeny of the Metazoa: evaluating alternative phylogenetic positions of Platyhelminthes, Nemertea, and Gnathostomulida, with a critical reappraisal of cladistic characters. Contributions to Zoology, 73, 3–163.
Jenner, R. A., & Scholtz, G. (2005). Playing another round of metazoan phylogenetics: historical epistemology, sensitivity analysis, and the position of Arthropoda within the Metazoa on the basis of morphology. In S. Koenemann, & R. A. Jenner (Eds.), Crustacea and arthropod relationships. Crustacean Issues, 16, 355–385.
Jensen, S., Droser, M. L., & Gehling, J. G. (2005). Trace fossil preservation and the early evolution of animals. Palaeogeography, Palaeoclimatology, Palaeoecology, 220, 19–29.
Jondelius, U., Ruiz-Trillo, I., Baguñà, J., & Riutort, M. (2002). The Nemertodermatida are basal bilaterians and not members of the Platyhelminthes. Zoologica Scripta, 31, 201–215.
Kapp, H. (2000). The unique embryology of Chaetognatha. Zoologischer Anzeiger, 239, 263–266.
Kaul, S., & Stach, T. (2010). Ontogeny of the collar cord: neurulation in the hemichordate Saccoglossus kowalevskii. Journal of Morphology, 271, 1240–1259.
Kluge, A. G., & Farris, J. S. (1969). Quantitative phyletics and the evolution of anurans. Systematic Zoology, 18, 1–32.
Kristensen, R. M. (2003). Comparative morphology: do the ultrastructural investigations of Loricifera and Tardigrada support the clade Ecdysozoa? In A. Legakis, S. Sfenthourakis, R. Polymeni, & M. Thessalou-Legaki (Eds.), The new panorama of animal evolution. Proceedings of the 18th International Congress of Zoology (pp. 467–477). Sofia: Pensoft.
Kristensen, R. M., & Funch, P. (2000). Micrognathozoa: a new class with complicated jaws like those of Rotifera and Gnathostomulida. Journal of Morphology, 246, 1–49.
Kusche, K., Bangel, N., Mueller, C., Hildebrandt, J.-P., & Weber, W.-M. (2005). Molecular cloning and sequencing of the Na+/K+-ATPase α-subunit of the medical leech Hirudo medicinalis (Annelida)—implications for modeling protostomian evolution. Journal of Zoological Systematics and Evolutionary Research, 43, 339–342.
Kusserow, A., Pang, K., Sturm, C., Hrouda, M., Lentfer, J., Schmidt, H. A., et al. (2005). Unexpected complexity of the Wnt gene family in a sea anemone. Nature, 433, 156–160.
Lake, J. A. (1990). Origin of the metazoa. Proceedings of the National Academy of Sciences of the USA, 87, 763–766.
Lartillot, N., & Philippe, H. (2008). Improvement of molecular phylogenetic inference and phylogeny of Bilateria. Proceedings of the Royal Society, Series B, 363, 1463–1472.
Lemmons, D., Fritzenwanker, J. H., Gerhart, J., Lowe, C. J., & McGinnis, W. (2010). Co-option of an anteroposterior head axis patterning system for proximodistal patterning of appendages in early bilaterian evolution. Developmental Biology, 344, 358–362.
Longhorn, S. J., Foster, P. G., & Vogler, A. P. (2007). The nematode-arthropod clade revisited: phylogenomic analyses from ribosomal protein genes misled by shared evolutionary biases. Cladistics, 23, 130–144.
Love, G. D., Grosjean, E., Stalvies, C., Fike, D. A., Grotzinger, J. P., Bradley, A. S., et al. (2009). Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature, 457, 718–722.
Lundin, K. (1998). The epidermal ciliary rootlets of Xenoturbella bocki (Xenoturbellida) revisited: new support for a possible kinship with the Acoelomorpha (Platyhelminthes). Zoologica Scripta, 27, 263–270.
Lundin, K. (2001). Degenerating epidermal cells in Xenoturbella bocki (phylum uncertain), Nemertodermatida and Acoela (Platyhelminthes). Belgian Journal of Zoology, 131, 153–157.
Lüter, C. (2000). Ultrastructure of larval and adult setae of Brachiopoda. Zoologischer Anzeiger, 239, 75–90.
Maas, A., Waloszek, D., Haug, J. T., & Müller, K. J. (2009). Loricate larvae (Scalidophora) from the Middle Cambrian of Australia. Memoirs of the Association of Australasian Palaeontologists, 37, 281–302.
Mallatt, J., & Winchell, C. J. (2002). Testing the new animal phylogeny: first use of combined large-subunit and small-subunit rRNA gene sequences to classify the protostomes. Molecular Biology and Evolution, 19, 289–301.
Mallatt, J., & Chen, J.-Y. (2003). Fossil sister group of craniates: predicted and found. Journal of Morphology, 258, 1–31.
Mallatt, J., & Giribet, G. (2006). Further use of nearly complete 28S and 18S rRNA genes to classify Ecdysozoa: 37 more arthropods and a kinorhynch. Molecular Biology and Evolution, 40, 772–794.
Mallatt, J., & Winchell, C. J. (2007). Ribosomal RNA genes and deuterostome phylogeny revisited: more cyclostomes, elasmobranchs, reptiles, and a brittle star. Molecular Phylogenetics and Evolution, 43, 1005–1022.
Mallatt, J., Garey, J. R., & Shultz, J. W. (2004). Ecdysozoan phylogeny and Bayesian inference: first use of nearly complete 28S and 18S rRNA gene sequences to classify the arthropods and their kin. Molecular Phylogenetics and Evolution, 31, 179–191.
Mallatt, J., Craig, C. W., & Yoder, M. J. (2010). Nearly complete rRNA genes assembled from across the metazoan animals: effects of more taxa, a structure-based alignment, and paired-sites evolutionary models on phylogenetic reconstruction. Molecular Phylogenetics and Evolution, 55, 1–17.
Marlétaz, F., Martin, E., Perez, Y., Papillon, D., Caubit, X., Lowe, C. J., et al. (2006). Chaetognath phylogenomics: a protostome with deuterostome-like development. Current Biology, 16, R578.
Martindale, M. Q., Pang, K., & Finnerty, J. R. (2004). Investigating the origins of triploblasty: ‘mesodermal’ gene expression in a diploblastic animal, the sea anemone Nematostella vectensis (phylum, Cnidaria; class, Anthozoa). Development, 131, 2463–2474.
Maslakova, S. A., Martindale, M. Q., & Norenburg, J. L. (2004a). Fundamental properties of the spiralian developmental program are displayed by the basal nemertean Carinoma tremaphoros (Palaeonemertea, Nemertea). Developmental Biology, 267, 342–360.
Maslakova, S. A., Martindale, M. Q., & Norenburg, J. L. (2004b). Vestigial prototroch in a basal nemertean, Carinoma tremaphoros (Nemertea; Palaeonemertea). Evolution & Development, 6, 219–226.
Matus, D. Q., Copley, R. R., Dunn, C. W., Hejnol, A., Eccleston, H., Halanych, K. M., et al. (2006). Broad taxon and gene sampling indicate that chaetognaths are protostomes. Current Biology, 16, R575–R576.
Meusemann, K., von Reumont, B. M., Simon, S., Roeding, F., Strauss, S., Kuck, P., et al. (2010). A phylogenomic approach to resolve the arthropod tree of life. Molecular Biology and Evolution, 27, 2451–2464.
Meyer, N. P., Boyle, M. J., Martindale, M. Q., & Seaver, E. C. (2010). A comprehensive fate map by intracellular injection of identified blastomeres in the marine polychaete Capitella teleta. EvoDevo, 1/8.
Mwinyi, A., Meyer, A., Bleidorn, C., Lieb, B., Bartolomaeus, T., & Podsiadlowski, L. (2009). Mitochondrial genome sequence and gene order of Sipunculus nudus give additional support for an inclusion of Sipuncula into Annelida. BMC Genomics, 10, 27.
Neuhaus, B., & Higgins, R. P. (2002). Ultrastructure, biology, and phylogenetic relationships of Kinorhyncha. Integrative and Comparative Biology, 42, 619–632.
Neves, R. C., Cunda, M. R., Kristensen, R. M., & Wanninger, A. (2010). Expression of synapsin and co-localization with serotonin and Rfamide-like immunoreactivity in the nervous system of the chordoid larva of Symbion pandora (Cycliophora). Invertebrate Biology, 129, 17–26.
Nickel, M. (2010). Evolutionary emergence of synaptic nervous systems: what can we learn from the non-synaptic, nerveless Porifera? Invertebrate Biology, 129, 1–16.
Nielsen, C. (1995). Animal evolution. Interrelationships of the living phyla (1st ed.). Oxford: Oxford University Press.
Nielsen, C. (2001). Animal evolution. Interrelationships of the living phyla (2nd ed.). Oxford: Oxford University Press.
Nielsen, C. (2010). After all: Xenoturbella is an acoelomorph! Evolution & Development, 12, 241–243.
Nielsen, C., Scharff, N., & Eibye-Jacobsen, D. (1995). Cladistic analysis of the animal kingdom. Biological Journal of the Linnean Society, 57, 385–410.
Paps, J., Baguñà, J., & Riutort, M. (2009a). Lophotrochozoa internal phylogeny: new insights from an up-to-date analysis of nuclear ribosomal genes. Proceedings of the Royal Society, Series B, 276, 1245–1254.
Paps, J., Baguñà, J., & Riutort, M. (2009b). Bilaterian phylogeny: a broad sampling of 13 nuclear genes provides a new Lophotrochozoa phylogeny and supports a paraphyletic basal Acoelomorpha. Molecular Biology and Evolution, 26, 2397–2406.
Pardos, F. (1988). Fine structure and function of pharynx cilia in Glossobalanus minutus Kowalewsky (Enteropneusta). Acta Zoologica, 69, 1–12.
Park, J.-K., Rho, H. S., Kristensen, R. M., Kim, W., & Giribet, G. (2006). First molecular data on the phylum Loricifera—an investigation into the phylogeny of Ecdysozoa with emphasis on the positions of Loricifera and Priapulida. Zoological Science, 23, 943–954.
Passamaneck, Y. J., Furchheim, N., Hejnol, A., Martindale, M. Q., & Lüter, C. (2011). Ciliary photoreceptors in the cerebral eyes of a protostome larva. EvoDevo, 2: 6.
Pedersen, K. J., & Pedersen, L. R. (1986). Fine structural observations on the extracellular matrix (ECM) of Xenoturbella bocki Westblad, 1949. Acta Zoologica, 67, 103–113.
Pedersen, K. J., & Pedersen, L. R. (1988). Ultrastructural observations on the epidermis of Xenoturbella bocki Westblad, 1949, with a discusion of epidermal cytoplasmic filament systems of invertebrates. Acta Zoologica, 69, 231–246.
Peel, J. S. (2010). A corset-like fossil from the Cambrian Sirius Passet Lagerstätte of North Greenland and its implications for cycloneuralian evolution. Journal of Paleontology, 84, 332–340.
Perseke, M., Hankeln, T., Weich, B., Fritzsch, G., Stadler, P. F., Israelsson, O., et al. (2007). The mitochondrial DNA of Xenoturbella bocki: genomic architecture and phylogenetic analysis. Theory in Biosciences, 126, 35–42.
Peterson, K. J., & Eernisse, D. J. (2001). Animal phylogeny and the ancestry of bilaterians: inference from morphology and 18S rDNA gene sequences. Evolution & Development, 3, 170–205.
Peterson, K. J., Cotton, J. A., Gehling, J. G., & Pisani, D. (2008). The Ediacaran emergence of bilaterians: congruence between the genetic and the geological fossil records. Philosophical Transactions of the Royal Society / Biological Sciences, 1496, 1435–1444.
Petrov, N. B., & Vladychenskaya, N. S. (2005). Phylogeny of molting protostomes (Ecdysozoa) as inferred from 18S and 28S rRNA gene sequences. Molecular Biology, 39, 503–513.
Philip, G. K., Creevey, C. J., & McInerney, J. O. (2005). The Opisthokonta and the Ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the Coelomata than Ecdysozoa. Molecular Biology and Evolution, 22, 1175–1184.
Philippe, H., Snell, E. A., Bapteste, E., Lopez, P., Holland, P. W. H., & Casane, D. (2004). Phylogenomics of eukaryotes: impact of missing data on large alignments. Molecular Biology and Evolution, 21, 1740–1752.
Philippe, H., Lartillot, N., & Brinkmann, H. (2005). Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Molecular Biology and Evolution, 22, 1246–1253.
Philippe, H., Brinkmann, H., Martinez, P., Riutort, M., & Baguñà, J. (2007). Acoel flatworms are not Platyhelminthes: evidence from phylogenomics. PloS ONE, 8, e717.
Philippe, H., Derelle, R., Lopez, P., Pick, K., Borchianelli, C., Boury-Esnault, N., et al. (2009). Phylogenomics revives traditional views on deep animal relationships. Current Biology, 19, 706–712.
Philippe, H., Brinkmann, H., Copley, R. R., Moroz, L. L., Nakano, H., Poustka, A. J., et al. (2011). Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature, 470, 255–258.
Pick, K. S., Philippe, H., Schreiber, F., Erpenbeck, D., Jackson, D. J., Wrede, P., et al. (2010). Improved phylogenomic taxon sampling noticeably affects non-bilaterian relationships. Molecular Biology and Evolution, 27, 1983–1987.
Pilato, G., Binda, M. G., Biondi, O., D’Urso, V., Lisi, O., Marletta, A., et al. (2005). The clade Ecdysozoa, perplexities and questions. Zoologischer Anzeiger, 244, 43–50.
Podar, M., Haddock, S. H. D., Sogin, M. L., & Harbison, G. R. (2001). A molecular phylogenetic framework for the phylum Ctenophora using 18S rRNA genes. Molecular Phylogenetics and Evolution, 21, 218–230.
Prendini, L. (2001). Species or supraspecific taxa as terminals in cladistic analysis? Groundplans versus exemplars revisited. Systematic Biology, 50, 290–300.
Raff, R. A., Field, K. G., Olsen, G. J., Giovannoni, S. J., Lane, D. J., Ghiselin, M. T., et al. (1989). Metazoan phylogeny based on analysis of 18S ribosomal RNA. In B. Fernholm, K. Bremer, & H. Jörnvall (Eds.), The hierarchy of life (pp. 247–260). Amsterdam: Elsevier Science BV.
Raikova, O. I., Reuter, M., Jondelius, U., & Gustafsson, M. K. S. (2000). An immunocytochemical and ultrastructural study of the nervous and muscular systems of Xenoturbella westbladi (Bilateria inc. sed.). Zoomorphology, 120, 107–118.
Raikova, O. I., Reuter, M., Gustafsson, M. K. S., Maule, A. G., Halton, D. W., & Jondelius, U. (2004a). Basiepidermal nervous system in Nemertoderma westbladi (Nemertodermatida): GYIRFamide immunoreactivity. Zoology (Jena), 107, 75–86.
Raikova, O. I., Reuter, M., Gustafsson, M. K. S., Maule, A. G., Halton, D. W., & Jondelius, U. (2004b). Evolution of the nervous system in Paraphanostoma (Acoela). Zoologica Scripta, 33, 71–88.
Rieger, R. M. (1980). A new group of interstitial worms, Lobatocerebridae nov. fam. (Annelida) and its significance for metazoan phylogeny. Zoomorphology, 95, 41–84.
Rieger, R. M. (1991). Jennaria pulchra, nov.gen. nov.spec., eine den psammobionten Anneliden nahestehende Gattung aus dem Küstengrundwasser von North Carolina. Berichte des Naturwissenschaftlich-Medizinischen Vereins in Innsbruck, 78, 203–215.
Roeding, F., Hagner-Holler, S., Ruhberg, H., Ebersberger, I., Haeseler, A., Kube, M., et al. (2007). EST sequencing of Onychophora and phylogenomic analysis of Metazoa. Molecular Biology and Evolution, 45, 942–951.
Roeding, F., Borner, J., Kube, M., Klages, M., Reinhardt, R., & Burmester, T. (2009). A 454 sequencing approach for large scale phylogenomic analysis of the common emperor scorpion (Pandinus imperator). Molecular Phylogenetics and Evolution, 53, 826–834.
Rogozin, I. B., Wolf, Y. I., Carmel, L., & Koonin, E. V. (2007). Ecdysozoan clade rejected by genome-wide analysis of rare amino acid replacements. Molecular Biology and Evolution, 24, 1080–1090.
Rokas, A., Krüger, D., & Carroll, S. B. (2005). Animal evolution and the molecular signature of radiations compressed in time. Science, 310, 1933–1938.
de Rosa, R., Grenier, J. K., Andreeva, T., Cook, C. E., Adoutte, A., Akam, M., et al. (1999). Hox genes in brachiopods and priapulids and protostome evolution. Nature, 399, 772–776.
Rota-Stabelli, O., Campbell, L., Brinkmann, H., Edgecombe, G. D., Longhorn, S. J., Peterson, K. J., et al. (2010a). A congruent solution to arthropod phylogeny: phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proceedings of the Royal Society B: Biological Sciences, 278, 298–306.
Rota-Stabelli, O., Kayal, E., Gleeson, D., Daub, J., Boore, J. L., Telford, M. J., et al. (2010b). Ecdysozoan mitogenomics: evidence for a common origin of the legged invertebrates, the Panarthropoda. Genome Biology and Evolution, 2, 425–440.
Rothe, B. H., & Schmidt-Rhaesa, A. (2009). Architecture of the nervous system in two Dactylopoda species (Gastrotricha, Macrodasyida). Zoomorphology, 128, 227–246.
Roule, L. (1891). Considerations sur l’embranchement des Trochozoaires. Annales des Sciences Naturelles (Zoologie), 7me Série, 11, 121–178.
Rouse, G. W. (1999). Trochophore concepts: ciliary bands and the evolution of larvae in spiralian Metazoa. Biological Journal of the Linnean Society, 66, 411–464.
Rouse, G. W., & Pleijel, F. (2007). Annelida. Zootaxa, 1668, 245–264.
Ruiz-Trillo, I., Riutort, M., Littlewood, D. T. J., Herniou, E. A., & Baguñà, J. (1999). Acoel flatworms: earliest extant bilaterian metazoans, not members of Platyhelminthes. Science, 283, 1919–1923.
Ruiz-Trillo, I., Paps, J., Loukota, M., Ribera, C., Jondelius, U., Baguñà, J., et al. (2002). A phylogenetic analysis of myosin heavy chain type II sequences corroborates that Acoela and Nemertodermatida are basal bilaterians. Proceedings of the National Academy of Sciences of the USA, 99, 11246–11251.
Ruppert, E. E. (1991). Gastrotricha. In F. W. Harrison & E. E. Ruppert (Eds.), Microscopic anatomy of invertebrates, volume 4: Aschelminthes (pp. 41–109). New York: Wiley-Liss Inc.
Ryan, J. F., Pang, K., NISC Comparative Sequencing Program, Mullikin, J. C., Martindale, M. Q., & Baxevanis, A. D. (2010). The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. EvoDevo, 1/9.
Sanderson, M. J. (2002). Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Molecular Biology and Evolution, 19, 101–109.
Sanderson, M. J. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics, 19, 301–302.
Sanderson, M. J. (2008). Phylogenetic signal in the eukaryotic tree of life. Science, 321, 121–123.
Schierwater, B., Eitel, M., Jakob, W., Osigus, H.-J., Hadrys, H., Dellaporta, S. L., et al. (2009). Concatenated analysis sheds light on early metazoan evolution and fuels a modern “Urmetazoan” hypothesis. PLoS Biology, 7, 36–44.
Schleip, W. (1929). Die Determination der Primitiventwicklung. Leipzig: Akademische Verlagsgesellschaft.
Schmidt-Rhaesa, A. (2004). Ecdysozoa versus Articulata. Sitzungsberichte der Gesellschaft Naturforschender Freunde zu Berlin, 43, 35–49.
Schmidt-Rhaesa, A. (2006). Perplexities concerning the Ecdysozoa: a reply to Pilato et al. Zoologischer Anzeiger, 244, 205–208.
Schmidt-Rhaesa, A. (2007). The evolution of organ systems. Oxford: Oxford University Press.
Schmidt-Rhaesa, A., Bartolomaeus, T., Lemburg, C., Ehlers, U., & Garey, J. R. (1998). The position of the Arthropoda in the phylogenetic system. Journal of Morphology, 238, 263–285.
Scholtz, G. (2002). The Articulata hypothesis—or what is a segment? Organisms Diversity and Evolution, 2, 197–215.
Scholtz, G. (2003). Is the taxon Articulata obsolete? Arguments in favour of a close relationship between annelids and arthropods. In A. Legakis, S. Sfenthourakis, R. Polymeni, & M. Thessalou-Legaki (Eds.), The new panorama of animal evolution. Proceedings of the 18th International Congress of Zoology (pp. 489–501). Sofia: Pensoft.
Schram, F. R. (1991). Cladistic analysis of metazoan phyla and the placement of fossil problematica. In A. M. Simonetta & S. Conway-Morris (Eds.), The early evolution of Metazoa and the significance of problematic taxa (pp. 35–46). Cambridge: Cambridge University Press.
Schram, F. R., & Ellis, W. N. (1994). Metazoan relationships: a rebuttal. Cladistics, 10, 331–337.
Seaver, E. C. (2003). Segmentation: mono- or polyphyletic? International Journal of Developmental Biology, 47, 583–595.
Sempere, L. F., Cole, C. N., McPeek, M. A., & Peterson, K. J. (2006). The phylogenetic distribution of metazoan microRNAs: insights into evolutionary complexity and constraint. Journal of Experimental Zoology, Part B: Molecular and Developmental Evolution, 306, 575–588.
Sempere, L. F., Martinez, P., Cole, C., Baguñà, J., & Peterson, K. J. (2007). Phylogenetic distribution of microRNAs supports the basal position of acoel flatworms and the polyphyly of Platyhelminthes. Evolution & Development, 9, 409–415.
Shu, D. G., Conway Morris, S., Han, J., Chen, L., Zhang, X. L., Zhang, Z. F., et al. (2001). Primitive deuterostomes from the Chengjiang Lagerstätte (Lower Cambrian, China). Nature, 414, 419–424.
Shu, D. G., Conway Morris, S., Zhang, Z. F., Liu, J. N., Han, J., Chen, L., et al. (2003). A new species of yunnanozoan with implications for deuterostome evolution. Science, 299, 1380–1384.
Shu, D.-G., Conway Morris, S., Han, J., Zhang, Z.-F., & Liu, J.-N. (2004). Ancestral echinoderms from the Chengjiang deposits of China. Nature, 430, 422–428.
Shu, D.-G., Conway Morris, S., Zhang, Z.-F., & Han, J. (2010). The earliest history of the deuterostomes: the importance of the Chengjiang Fossil-Lagerstätte. Proceedings of the Royal Society, Series B, 277, 165–174.
Siddall, M. E. (2009). Unringing the bell: metazoan phylogenomics and the partition bootstrap. Cladistics, 25, 1–9.
Slyusarev, G. S., & Kristensen, R. M. (2003). Fine structure of the ciliated cells and ciliary rootlets of Intoshia variabili (Orthonectida). Zoomorphology, 122, 33–39.
Smith, A. B. (2005). The pre-radial history of echinoderms. Geological Journal, 40, 255–280.
Smith, A. B., & Swalla, B. J. (2009). Deciphering deuterostome phylogeny: molecular, morphological, and palaeontological perspectives. In M. J. Telford & D. T. J. Littlewood (Eds.), Animal evolution: Genomes, fossils and trees (pp. 80–92). Oxford: Oxford University Press.
Sørensen, M. V. (2003). Further structures in the jaw apparatus of Limnognathia maerski (Micrognathozoa), with notes on the phylogeny of the Gnathifera. Journal of Morphology, 255, 131–145.
Sørensen, M. V., Funch, P., Willerslev, E., Hansen, A. J., & Olesen, J. (2000). On the phylogeny of the Metazoa in light of Cycliophora and Micrognathozoa. Zoologischer Anzeiger, 239, 297–318.
Sørensen, M. V., Hebsgaard, M. B., Heiner, I., Glenner, H., Willerslev, E., & Kristensen, R. M. (2008). New data from an enigmatic phylum: evidence from molecular sequence data supports a sister group relationship between Loricifera and Nematomorpha. Journal of Zoological Systematics and Evolutionary Research, 46, 231–239.
Sperling, E. A., Peterson, K. J., & Pisani, D. (2009). Phylogenetic signal-dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of Eumetazoa. Molecular Biology and Evolution, 26, 2261–2274.
Sperling, E. A., Vinther, J., Moy, V. N., Wheeler, B. M., Sémon, M., & Briggs, D. E. G. (2009). MicroRNAs resolve an apparent conflict between annelid systematics and their fossil record. Proceedings of the Royal Society B / Biological Sciences, 276, 4315–4322.
Sperling, E. A., Robinson, J. M., Pisani, D., & Peterson, K. J. (2010). Where’s the glass? Biomarkers, molecular clocks and microRNAs suggest a 200-Myr missing Precambrian fossil record of siliceous sponge spicules. Geobiology, 8, 24–36.
Srivastava, M., Begovic, E., Chapman, J., Putnam, N. H., Hellsten, U., Kawashima, T., et al. (2008). The Trichoplax genome and the nature of placozoans. Nature, 454, 955–960.
Srivastava, M., Simakov, O., Chapman, J., Fahey, B., Gauthier, M. E., et al. (2010). The Amphimedon queenslandica genome and the evolution of animal complexity. Nature, 466, 720–726.
Struck, T. H., & Fisse, F. (2008). Phylogenetic position of Nemertea derived from phylogenomic data. Molecular Biology and Evolution, 25, 728–736.
Struck, T. H., Schult, N., Kusen, T., Hickman, E., Bleidorn, C., McHugh, D., et al. (2007). Annelid phylogeny and the status of Sipuncula and Echiura. BMC Evolutionary Biology, 7, 11.
Telford, M. J. (2006). Animal phylogeny. Current Biology, 16, R981–R985.
Telford, M. J., Bourlat, S. J., Economou, A., Papillon, D., & Rota-Stabelli, O. (2008). The evolution of the Ecdysozoa. Philosophical Transactions of the Royal Society, Series B, 363, 1529–1537.
Todaro, M. A., Telford, M. J., Lockyer, A. E., & Littlewood, D. T. (2006). Interrelationships of the Gastrotricha and their place among the Metazoa inferred from 18S rRNA genes. Zoologica Scripta, 35, 251–259.
Townsend, J. P. (2007). Profiling phylogenetic informativeness. Systematic Biology, 56, 222–231.
Valentine, J. W. (2004). On the origin of phyla. Chicago: The University of Chicago Press.
Voigt, O., Collins, A. G., Porello, S., Pearse, V. B., & Schierwater, B. (2004). Placozoa—no longer a phylum of one. Current Biology, 14, R944–R945.
Wallace, R. L., Ricci, C., & Melone, G. (1996). A cladistic analysis of pseudocoelomate (aschelminth) morphology. Invertebrate Biology, 115, 104–112.
Wallberg, A. (2009). The dawn of a new age. Interrelationships of Acoela and Nemertodermatida and the early evolution of Bilateria. Doctoral thesis. Uppsala: Uppsala University.
Wallberg, A., Curini-Galletti, M., Ahmadzadeh, A., & Jondelius, U. (2007). Dismissal of Acoelomorpha: Acoela and Nemertodermatida are separate early bilaterian clades. Zoologica Scripta, 36, 509–523.
Wanninger, A. (2008). Comparative lophotrochozoan neurogenesis and larval neuroanatomy: recent advances from previously neglected taxa. Acta Biologica Hungarica, 59(Supplement), 127–136.
Webster, B. L., Copley, R. R., Jenner, R. A., Mackenzie-Dodds, J. A., Bourlat, S. J., Rota-Stabelli, O., et al. (2006). Mitogenomics and phylogenomics reveal priapulid worms as extant models for the ancestral Ecdysozoan. Evolution & Development, 8, 502–510.
Webster, B. L., Mackenzie-Dodds, J. A., Telford, M. J., & Littlewood, D. T. J. (2007). The mitochondrial genome of Priapulus caudatus Lamarck (Priapulida: Priapulidae). Gene, 389, 96–105.
Westblad, E. (1949). Xenoturbella bocki n. g., n. sp., a peculiar, primitive turbellarian type. Arkiv för Zoologi, 1, 3–29.
Wheeler, B. M., Heimberg, A. M., Moy, V. N., Sperling, E. A., Holstein, T. W., Heber, S., et al. (2009). The deep evolution of metazoan microRNAs. Evolution & Development, 11, 50–68.
Winchell, C. J., Sullivan, J., Cameron, C. B., Swalla, B. J., & Mallatt, J. (2002). Evaluating hypotheses of deuterostome phylogeny and chordate evolution with new LSU and SSU ribosomal DNA data. Molecular Biology and Evolution, 19, 762–776.
Winnepenninckx, B., Backeljau, T., & De Wachter, R. (1995a). Phylogeny of protostome worms derived from 18S rRNA sequences. Molecular Biology and Evolution, 12, 641–649.
Winnepenninckx, B., Backeljau, T., Mackey, L. Y., Brooks, J. M., De Wachter, R., Kumar, S., et al. (1995b). 18S rRNA data indicate that Aschelminthes are polyphyletic in orgin and consist of at least three distinct clades. Molecular Biology and Evolution, 12, 1132–1137.
Witek, A., Herlyn, H., Ebersberger, I., Welch, D. B. M., & Hankeln, T. (2009). Support for the monophyletic origin of Gnathifera from phylogenomics. Molecular Phylogenetics and Evolution, 53, 1037–1041.
Wolf, Y. I., Rogozin, I. B., & Koonin, E. V. (2004). Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Research, 14, 29–36.
Worsaae, K., & Rouse, G. W. (2008). Is Diurodrilus an annelid? Journal of Morphology, 269, 1426–1455.
Xiao, S., & Laflamme, M. (2009). On the eve of animal radiation: phylogeny, ecology and evolution of the Ediacaran biota. Trends in Ecology and Evolution, 24, 31–40.
Zrzavý, J. (2001). Ecdysozoa versus Articulata: clades, artifacts, prejudices. Journal of Zoological Systematics and Evolutionary Research, 39, 159–163.
Zrzavý, J. (2003). Gastrotricha and metazoan phylogeny. Zoologica Scripta, 32, 61–81.
Zrzavý, J., Mihulka, S., Kepka, P., Bezdek, A., & Tietz, D. (1998). Phylogeny of the Metazoa based on morphological and 18S ribosomal DNA evidence. Cladistics, 14, 249–285.
Zrzavý, J., Hypša, V., & Tietz, D. F. (2001). Myzostomida are not annelids: molecular and morphological support for a clade of animals with anterior sperm flagella. Cladistics, 17, 170–198.
Acknowledgements
Rudolf Meier requested this review and handled the paper for the journal. We thank him, Olaf Bininda-Emonds, Andreas Schmidt-Rhaesa and a second, anonymous reviewer for providing comments that helped to improve an earlier version of this manuscript. Claus Nielsen and Akiko Okusu kindly made the line drawings used in Figure 1 available, which improved upon earlier versions by Miquel A. Arnedo. The Carlsberg Foundation provided funding for collecting in Greenland (grant 2009_01_0053). This work is supported by the National Science Foundation under the AToL program (grants EF05-31757, EF05-31558 and EF05-31677).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 70 kb)
Rights and permissions
About this article
Cite this article
Edgecombe, G.D., Giribet, G., Dunn, C.W. et al. Higher-level metazoan relationships: recent progress and remaining questions. Org Divers Evol 11, 151–172 (2011). https://doi.org/10.1007/s13127-011-0044-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-011-0044-4