
Abstract
Diabetes is an independent risk factor for atrial fibrillation (AF). This study aimed to elucidate the pathophysiology of diabetes-related AF from the perspective of the gut microbial metabolite trimethylamine N-oxide (TMAO). In the present study, male rats received either a normal diet to serve as the control group or a high-fat diet/streptozotocin to induce type 2 diabetes mellitus. Then, diabetic rats were divided into two groups based on the presence or absence of 3,3-dimethyl-1-butanol (DMB, a specific TMAO inhibitor) in drinking water: the diabetic cardiomyopathy (DCM) group and the DCM + DMB group. Eight weeks later, compared with control rats, rats in the DCM group exhibited gut microbiota dysbiosis and systemic TMAO elevation. The inflammatory cytokines IL-1β, IL-6, and TNF-α were markedly increased in the atria of rats in the DCM group. Downregulated expression of connexin 40 and lateralized distribution of connexin 43 were also observed in the atria of DCM rats. AF inducibility was significantly higher in DCM rats than in control rats. Furthermore, DMB treatment effectively ameliorated atrial inflammation and connexin remodeling while markedly reducing plasma TMAO levels. DMB treatment also decreased the vulnerability of diabetic rats to AF. In conclusion, TMAO might promote atrial inflammation and connexin remodeling in the development of diabetes, which may play a key role in mediating diabetes-related AF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is the most common sustained tachycardia and has become a major social and economic burden worldwide [28]. Many risk factors, such as obesity and hypertension, contribute to the development of AF. Among these contributors, diabetes is verified to be an independent risk factor for AF [21]. The Framingham Heart Study demonstrated that diabetes was associated with an increased risk of developing AF, with an odds ratio of 1.4 in men and 1.6 in women [2]. A meta-analysis also showed that patients with diabetes had a 40% greater risk of developing AF than patients without diabetes [13]. In addition, numerous studies have verified that diabetic patients with AF are at higher risk of all-cause death, cardiovascular mortality, and heart failure than those without AF [8]. Therefore, AF should be regarded as a marker of poor prognosis in diabetic patients, and appropriate approaches should be taken to target the progression of AF in diabetic patients.
Despite numerous basic and clinical studies, the pathophysiology underlying AF is still not completely understood. Moreover, the mechanism of diabetes-related AF is even more confusing. Recent studies have reported that chronic inflammation may play an important role in cardiac arrhythmia. Lazzerini et al. found that chronic inflammation activated by rheumatoid arthritis promoted malignant ventricular arrhythmias and atrial arrhythmias by affecting cardiac electrophysiology directly or aggravating cardiac pathological remodeling indirectly [15]. Similarly, several studies have demonstrated that activated inflammation facilitated postoperative AF, while inhibition of the inflammatory response reduced the incidence of postoperative AF [33]. Type 2 diabetes mellitus (T2DM) is now recognized as an inflammatory disorder [7, 20]. Moreover, there are growing lines of evidence indicating that chronic low-grade myocardial inflammation is a notable characteristic in the diabetic heart and plays an important role in the development of diabetic cardiomyopathy [19, 20]. Clinical studies have reported that diabetes is accompanied by elevated levels of systemic inflammatory mediators [9]. Basic studies also confirmed that various proinflammatory pathways were activated in the myocardium of diabetic rats [10]. Therefore, chronic cardiac inflammation might be a key bridge between diabetes and arrhythmia.
Currently, numerous studies on the gut microbiota have emerged. Recent studies have reported that abnormalities in the gut microbiota and its metabolites are closely related to systemic inflammation [4]. Among these gut microbiota-derived metabolites, trimethylamine N-oxide (TMAO) was found to promote chronic inflammation in a variety of disease models. Chen et al. found that TMAO can induce inflammation by activating the NLR family pyrin domain containing 3 inflammasomes [3]. Similarly, Seldin et al. reported that TMAO was able to promote inflammation through mitogen-activated protein kinase and nuclear factor-κB signaling [26]. Intriguingly, elevated TMAO levels were also found in diabetes models [5]. Clinical studies have demonstrated that diabetic patients, especially those with cardiomyopathy, showed disordered gut microbiota and significantly increased TMAO levels [1] which may therefore contribute to the occurrence of cardiac inflammation and the induction of arrhythmia. However, the exact role of TMAO in diabetes-related arrhythmias remains speculative. Thus, in the present study, we constructed a type 2 diabetes (T2D) model by administering a high-fat diet with streptozotocin and used 3,3-dimethyl-1-butanol (DMB, an inhibitor of trimethylamine formation) to explore the role of TMAO in diabetes-related AF.
Materials and methods
Animal preparation
A total of thirty-six male Sprague–Dawley rats (120–140 g) were obtained from Nanjing Medical University Animal Center. All study protocols were approved by the Animal Ethics Committee of Nanjing Medical University (IACUC-2011003) and fully complied with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). All data were collected under blinded conditions during the whole experiment.
Study protocols
After 1 week of acclimatization, 24 rats were randomly assigned to the diabetes group, which was fed a high-fat diet (Research Diets, New Brunswick, New Jersey, USA) for 4 weeks, while the remaining 12 rats received a normal diet as the control group. All rats were fasted overnight on day 28. The next morning, rats in the diabetes group were intraperitoneally injected with low-dose streptozotocin (Sigma–Aldrich, St. Louis, MO, USA), which was dissolved in citrate buffer (0.1 mol/L, pH 4.5, 4 °C). Each rat was given streptozotocin at a dose of 35 mg/kg body weight. Rats in the control group were intraperitoneally injected with an equal volume of citrate buffer. One week later, 19 rats in the diabetes group developed hyperglycemia and insulin resistance, with fasting blood glucose ≥ 11.1 mmol/L and random blood glucose ≥ 16.7 mmol/L. Then, the 19 diabetic rats were randomly distributed into two groups: the diabetic cardiomyopathy (DCM) group (n = 10), which received a high-fat diet for 8 weeks, and the diabetic cardiomyopathy combined with 3,3-dimethyl-1-butanol (DCM + DMB) group (n = 9), which received a high-fat diet and 1% DMB in the drinking water for 8 weeks. Similarly, rats in the control group were randomly assigned to two groups: the control group (n = 6), which received a normal diet for 8 weeks, and the control combined with 3,3-dimethyl-1-butanol (control + DMB) group (n = 6), which received a normal diet and 1% DMB in the drinking water for 8 weeks. DMB, a structural analogue of choline, can effectively reduce circulating TMAO levels by inhibiting the formation of trimethylamine in the intestine. The dose of DMB was based on previous studies [30].
Echocardiography
At the end of the experiment, 6, 6, 7, and 7 rats survived in the control, control + DMB, DCM, and DCM + DMB groups, respectively. Echocardiography was performed to detect cardiac function with a Vevo2100 (VisualSonics, Toronto, Canada) system. Left ventricular systolic function was assessed by the ejection fraction (EF) and fractional shortening (FS). Left ventricular diastolic function was assessed by E (early diastolic inflow E-wave), A (late diastolic inflow A-wave), E/A (the ratio of the E to A waves), e’ velocities (early diastolic annular e’-wave), and E/e’ (the ratio of the E to e’ waves).
Electrophysiological study
An in vivo electrophysiological study was performed to determine the atrial electrical changes. After anaesthetization by intraperitoneal injection of 2% sodium pentobarbital, the rats were fixed on the operating table, and thoracotomy was performed to expose the right atrium and left atrium. Three needle electrodes each were placed on both of their upper limbs and the atrial sites. Programmed electrical stimulation was used to investigate the atrial effective refractory period (AERP) through a bipolar electrode. The AERP was measured under three basic cycle lengths (100 ms, 150 ms, and 200 ms), and the stimulus intensity was twice the pacing threshold. Eight basic stimuli (S1) were followed by an extra stimulus (S2). The S1S1 interval was progressively shortened by 10 ms, and the S1S2 interval was progressively shortened by 2 ms until no atrial electrical activity could be captured. The AERP referred to the longest S1S2 interval that failed to capture atrial activity. The AERP from different epicardial sites, including the high right atrium (HRA), high left atrium (HLA), and low left atrium (LLA), was recorded and used to calculate the AERP dispersion (AERPD), which referred to the difference between the longest AERP and the shortest AERP from three sites. AF inducibility was also detected by burst pacing (cycle length of 50 ms) for 1 s, repeated 5 times with a 30-s interval at different atrial sites. AF referred to rapid and irregular atrial excitations that were longer than 1 s. The inducibility of AF was the ratio of the number of AF inductions to the total number of stimuli.
Analysis of the gut microbiota
16S rDNA sequencing was used to detect the composition of the intestinal flora in fecal samples. Briefly, DNA from fecal samples was extracted using the E.Z.N.A. ®Stool DNA Kit (D4015, Omega, Inc., USA) according to the manufacturer’s instructions. The V3–V4 region of the 16S rDNA gene was amplified by PCR using the primers 341F (5ʹ-CCTACGGGNGGCWGCAG-3ʹ) and 805R (5ʹ -GACTACHVGGGTATCTAATCC-3ʹ). The PCR products were sequenced on an Illumina NovaSeq platform according to the manufacturer’s recommendations provided by LC-Bio. Then, the raw data were filtered, processed, and analyzed. The alpha diversity and beta diversity were both calculated by QIIME2 software and visualized by the R package (Version 3.5.2). Blast was used for sequence alignment, and the feature sequences were annotated with the SILVA database for each representative sequence.
Quantification of TMAO and biochemical analysis
Plasma samples were collected from venous blood and stored at − 80 °C until detection. The levels of plasma TMAO were quantified using stable isotope dilution liquid chromatography tandem mass spectrometry (LC–MS). The levels of blood glucose, triglyceride, and cholesterol in plasma samples were determined using a Roche Cobas 8000 modular analyzer system (Roche, Rotkreuz, Switzerland).
Enzyme-linked immunosorbent assay
The concentration of plasma fasting insulin was detected with a commercial ELISA kit (mlbio, Shanghai, China). The levels of plasma IL-1β and IL-6 were measured by using commercial ELISA kits according to the manufacturer’s instructions (MultiSciences, Hangzhou, China). The level of plasma TNF-α was measured with a commercial ELISA kit (Thermo Fisher Scientific, Waltham, MA, USA).
Histopathology and immunohistochemistry
After sacrifice by intraperitoneal injection of an overdose of sodium pentobarbital, all rats were perfused with normal saline. Hearts were harvested and fixed with 4% paraformaldehyde for 48 h. After embedding and sectioning, atrial sections were used for histological analysis. Hematoxylin–eosin (HE) staining was performed to evaluate cardiac structural changes. To evaluate the expression of cytokines in the atrial myocardium, heart sections were stained with anti-interleukin-1β antibody (IL-1β, Servicebio, Wuhan, China), anti-interleukin-6 antibody (IL-6, Servicebio, Wuhan, China), and anti-tumor necrosis factor-alpha antibody (TNF-α, Servicebio, Wuhan, China) to assess the degree of inflammatory infiltration in the atrial myocardium. Five fields from each sample were randomly selected, and the integrated optical density (IOD) was calculated by Image-Pro Plus 6.0 software (National Institutes of Health, Bethesda, Maryland, USA).
Immunofluorescence
Immunofluorescence staining with an anti-connexin 40 antibody (Thermo Fisher Scientific, Waltham, MA, USA) and anti-connexin 43 antibody (Thermo Fisher Scientific, Waltham, MA, USA) was performed to detect the distribution of connexins in the atrium.
Western blotting
Western blotting was used to semiquantitatively analyze protein expression in the atrial tissues. Briefly, the atrial tissues were homogenized in mammalian tissue lysis buffer (Beyotime Biotechnology, Shanghai, China), and the protein concentrations were quantified using the BCA method (Beyotime Biotechnology, Shanghai, China). After electrophoresis, transfer, and blocking, these samples were probed with specific primary antibodies, including anti-connexin 40 (Cx40, Thermo Fisher Scientific, Waltham, MA, USA), anti-total connexin 43 (Cx43, Thermo Fisher Scientific, Waltham, MA, USA), anti-non-phosphorylated connexin 43 (np-Cx43, Thermo Fisher Scientific, Waltham, MA, USA), anti-phosphorylated connexin 43 (p-Cx43 Ser368, Cell Signaling Technology, Boston, Massachusetts, USA), anti-IL-1β (Servicebio, Wuhan, China), anti-IL-6 (Servicebio, Wuhan, China), and anti-TNF-α (Servicebio, Wuhan, China). Then, the membranes were incubated with the appropriate secondary antibodies. The protein bands were analyzed with ImageJ software (National Institutes of Health, Bethesda, Maryland, USA).
Statistical analysis
All statistical analyses in the present study were performed using GraphPad Prism 8 software (GraphPad Software, Inc. La Jolla, CA, USA), and all quantitative data are presented as the mean ± standard deviation (SD). Two-tailed unpaired t tests were used for two-group comparisons, while one-way ANOVA followed by the Newman–Keuls test was applied to multiple-group comparisons. Fisher’s exact test was used for qualitative data analysis. Statistical differences were considered significant if P < 0.05.
Results
Diabetic rats showed cardiac diastolic dysfunction and atrial pathological remodeling
In the present study, a T2D rat model was generated by long-term high-fat diet feeding combined with low-dose streptozotocin injection. After 3 months of high-fat diet feeding, diabetic rats developed obvious hyperglycemia and hyperinsulinemia (Fig. 1Aa, b). In addition, the levels of plasma triglyceride and total cholesterol were significantly higher in diabetic rats than in rats in the control group (Fig. 1Ac, d). These systemic metabolic disorders verified the successful establishment of a T2D model.

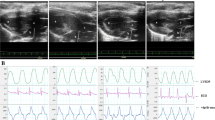
Diabetic rats showed cardiac diastolic dysfunction and atrial pathological remodeling. A Diabetic rats exhibited systemic metabolic disorders. (a) FBG. (b) FINS. (c) Triglyceride. (d) Cholesterol. B Diabetic rats showed diastolic dysfunction and atrial pathological remodeling. (a) Representative tracings of echocardiography and representative images of HE staining (magnification, 400 ×) in the atrium. (b) E/A ratio. (c) E/e’ ratio. (d) EF. (e) FS. (f) LAD. (g) LVPWd. (P < 0.05; n = 6, 6, 7, and 7 in the control, control + DMB, DCM, and DCM + DMB groups, respectively; five sections were randomly selected per sample.) *P < 0.05 vs. the control group; #P < 0.05 vs. the DCM group. FBG, fasting blood glucose; FINS, fasting insulin; EF, ejection fraction; FS, fractional shortening; LAD, left atrial diameter; LVPWd, left ventricular posterior wall diastolic thickness
Atrial function impairment is a common cardiovascular complication of diabetes. We next evaluated cardiac functional and structural changes in type 2 diabetic rats. As shown in Fig. 1B, echocardiography showed that diabetic rats had an enlarged left atrial diameter and thickened left ventricular posterior wall, as well as a reversed E/A ratio and increased E/e’ ratio, which collectively suggested that diabetic rats developed cardiac diastolic dysfunction and atrial structural changes. Furthermore, we detected atrial pathologic remodeling by HE staining. Diabetic rats exhibited cardiomyocyte hypertrophy and inflammatory cell infiltration in atrial tissues (Fig. 1B). In summary, diabetic rats developed diabetic cardiomyopathy at this stage.
Diabetic rats exhibited gut microbiota dysbiosis and plasma TMAO elevation
The composition and function of the gut microbiota are influenced by metabolic disorders in T2D. 16S rDNA sequencing was used to evaluate the changes in the intestinal flora in diabetic rats. Alpha diversity was applied to analyze species richness and species diversity within samples. Compared with rats in the control group, DCM rats had significantly decreased Chao1 richness, indicating that diabetes decreased intestinal microbial richness (Fig. 2Aa). The Simpson index and Shannon index represent species diversity and showed a decreasing trend in DCM rats (Fig. 2Ab, c). Beta diversity was used to analyze the microbial differences among the different groups. Principal component analysis showed significant variation in the microbiota composition between the control group and the DCM group (Fig. 2B).

Diabetic rats exhibited gut microbiota dysbiosis and plasma TMAO elevation. A Diabetic rats had decreased intestinal microbial richness. (a) The Chao1 index. (b) The Simpson index. (c) The Shannon index. B Diabetic rats had significant variation in the microbiota composition compared with control rats by principal component analysis. C The relative abundance of bacteria at the phylum level. D The relative abundance of bacteria at the order level. E Plasma TMAO level. (P < 0.05; n = 6, 6, 7, and 7 in the control, control + DMB, DCM, and DCM + DMB groups, respectively.) *P < 0.05 vs. the control group; #P < 0.05 vs. the DCM group. OTU, operational taxonomic units; TMAO, trimethylamine N-oxide
Next, we detected which bacterial taxa contributed to these differences. Figure 2C shows the intestinal microflora in different groups at the phylum level. The main microflora of rats in the control group were Firmicutes (62.16%), Bacteroidetes (33.74%), Proteobacteria (1.76%), Actinobacteria (1.14%), and other bacteria (1.19%). In contrast to the control group, DCM rats showed decreased Bacteroidetes (control 33.74% vs. DCM 3.62%), increased Firmicutes (control 62.16% vs. DCM 64.38%) and Proteobacteria (control 1.76% vs. DCM 26.94%), and an elevated Firmicutes/Bacteroidetes ratio. Furthermore, at the order level, we observed that diabetes significantly increased the relative abundances of Clostridiales (control 3.81% vs. DCM 18.96%, P < 0.05) and Desulfovibrionales (control 0.47% vs. DCM 22.86%, P < 0.05) while decreasing the relative abundance of Bacteroidales (control 32.83% vs. DCM 3.61%, P < 0.05; Fig. 2D).
The gut microbiota is closely associated with the level of plasma TMAO. Since we found that the relative abundance of flora related to TMAO production, including Clostridiales and Desulfovibrionales, was increased, we next detected plasma TMAO levels in the different groups. As illustrated in Fig. 2E, rats in the DCM group exhibited markedly increased plasma TMAO levels compared to levels in rats in the control group. All of the above results suggested that diabetic rats showed abnormal gut microbiota and elevated plasma TMAO levels.
Elevated TMAO levels promoted atrial inflammation in diabetic rats
Elevated plasma TMAO levels are associated with systemic inflammation. Therefore, we further measured the expression of inflammatory cytokines in the plasma and atrial tissues. Compared with rats in the control group, the inflammatory cytokines IL-1β, IL-6, and TNF-α were markedly increased in the plasma of rats in the DCM group (Fig. 3B). As shown in Fig. 3A, immunohistochemical staining indicated that the staining intensity of the inflammatory cytokines IL-1β, IL-6 and TNF-α was significantly increased in the atrium of rats in the DCM group (Fig. 3A). In line with this result, the relative expression of these inflammatory cytokines was also upregulated in the atrium of DCM rats, as evidenced by western blotting (Fig. 3C). These results collectively implied that atrial inflammation occurred in diabetic rats at this stage.

Elevated TMAO levels promoted atrial inflammation in diabetic rats. A Immunohistochemical staining of inflammatory cytokines in atrial tissues. (a) Representative images of IL-1β, IL-6, and TNF-α immunohistochemical staining (magnification, 400 ×) of rats from different groups. B ELISA analysis of inflammatory cytokines in the plasma. (a) The IL-1β levels. (b) The IL-6 levels. (c) The TNF-α levels. C Semiquantitative analysis of inflammatory cytokines in atrial tissues. (a) WB analysis of the protein levels of IL-1β, IL-6, and TNF-α in the atrium. (b) The relative levels of IL-1β calculated from the WB. (c) The relative levels of IL-6 calculated from the WB. (d) The relative levels of TNF-α calculated from the WB. (P < 0.05; n = 6, 6, 7, and 7 in the control, control + DMB, DCM, and DCM + DMB groups, respectively; five sections were randomly selected per sample.) *P < 0.05 vs. the control group; #P < 0.05 vs. the DCM group
To confirm the role of elevated TMAO in mediating inflammation, we applied a specific TMAO inhibitor, DMB, to suppress trimethylamine formation and reduce circulating TMAO levels [30]. After DMB treatment, although cardiac function and the gut microbiota did not show measurable improvement (Figs. 1B, 2A, B), the levels of plasma TMAO were significantly decreased in both the control + DMB group and the DCM + DMB group (Fig. 2E). On this basis, compared with DCM rats, DMB treatment effectively attenuated the expression of IL-1β, IL-6, and TNF-α in atrial tissues (Fig. 3). Thus, elevated TMAO levels play an important role in mediating atrial inflammation in diabetic rats.
Elevated TMAO levels facilitated connexin remodeling in diabetic rats
Cardiac inflammation can affect the distribution and expression of gap junction proteins, especially Cx43 and Cx40 [14]. As shown in Fig. 4A, immunofluorescence staining showed that Cx43 was mainly distributed in intercalated discs of atrial myocytes under normal circumstances. However, in DCM rats, Cx43 was predominantly localized at the cell periphery or cytoplasm and sporadically expressed at the intercalated discs of atrial myocytes. Similarly, compared with that in the control rats, apparent lateralization of Cx40 was also observed in the atrium of DCM rats. Moreover, the protein expression level of connexins was detected by western blotting. As shown in Fig. 4B, Cx40 was markedly decreased in the atrium of DCM rats. Although total Cx43 showed no obvious difference between the DCM group and the control group, the expression of nonphosphorylated Cx43 was markedly increased, while the expression of phosphorylated Cx43 was observably decreased in the DCM group. However, after DMB treatment, the distribution of gap junction proteins was markedly improved, the expression of Cx40 and phosphorylated Cx43 was significantly increased, and the expression of nonphosphorylated Cx43 was significantly decreased. Thus, DCM rats had an abnormal distribution and expression levels of gap junction proteins in the atrium, which was related to the elevated levels of TMAO.

Elevated TMAO levels facilitated connexin remodeling in diabetic rats. A Immunofluorescent staining of gap junction proteins in atrial tissues. (a) Representative images of Cx43 and Cx40 staining in the atrium (magnification, 400 ×). B Semiquantitative analysis of gap junction proteins in atrial tissues. (a) WB analysis of the protein levels of Cx40, total Cx43, nonphosphorylated Cx43, and phosphorylated Cx43 in the atrium. (b) The relative levels of Cx40 calculated from the WB. (c) The relative levels of total Cx43 calculated from the WB. (d) The relative levels of nonphosphorylated Cx43 calculated from the WB. (e) The relative levels of phosphorylated Cx43 calculated from the WB. (P < 0.05; n = 6, 6, 7, and 7 in the control, control + DMB, DCM, and DCM + DMB groups, respectively; five sections were randomly selected per sample.) * P < 0.05 vs. the control group; # P < 0.05 vs. the DCM group. Cx40, connexin 40; Cx43, connexin 43
Elevated TMAO levels increased the incidence of atrial fibrillation in diabetic rats
Gap junction proteins play a vital role in effective impulse propagation. Thus, we performed an electrophysiological study to assess atrial electrical changes among the groups. Figure 5A shows a schematic diagram of AF induced by burst pacing. As shown in Fig. 5F, AF inducibility was significantly higher in the DCM group than in the control group (control 16.67% vs. DCM 48.57%, P < 0.05). Although no significant differences in HRAERP, HLAERP, or LLAERP were found between the control group and the DCM group, AERPD was significantly higher in the DCM group than that in the control group (Fig. 5E). After DMB treatment, the AERPD was dramatically decreased, and AF inducibility was markedly reduced (DCM 48.57% vs. DCM + DMB 25.71%, P < 0.05), suggesting that abnormally elevated TMAO is an important cause of AF in diabetic rats.

Elevated TMAO levels increased the incidence of atrial fibrillation in diabetic rats. A Schematic diagram of the induction of AF. B HRAERP. C HLAERP. D LLAERP. E AERPD. F AF inducibility. (P < 0.05; n = 6, 6, 7, and 7 in the control, control + DMB, DCM, and DCM + DMB groups, respectively.) * P < 0.05 vs. the control group; # P < 0.05 vs. the DCM group. AF, atrial fibrillation; HRAERP, high right atrial effective refractory period; HLAERP, high left atrial effective refractory period; LLAERP, low left atrial effective refractory period; AERPD, atrial effective refractory period dispersion
Discussion
In the present study, we established a T2D model to investigate the role of TMAO in atrial fibrillation. The main findings were as follows: (1) Diabetic rats showed abnormal gut microbiota and increased TMAO levels. (2) Elevated systemic TMAO levels promoted inflammatory infiltration in atrial tissues. (3) Elevated systemic TMAO levels facilitated the remodeling of gap junction proteins in atrial tissues. (4) Atrial inflammation and connexin remodeling caused by elevated TMAO increased the susceptibility to AF in diabetic rats, and DMB treatment alleviated the progression of diabetes-related AF by reducing TMAO levels.
Animal models of type 2 diabetes mellitus
Currently, the prevalence of T2D is increasing at alarming rates. A plethora of animal models that simulate T2D have been established for exploring the pathogenesis or drug efficacy. In the present study, we constructed a T2D rat model by high-fat diet feeding for 3 months combined with low-dose streptozotocin injection. This model, which incorporates a high-fat diet to induce insulin resistance followed by low-dose streptozotocin to destroy a portion of the pancreatic β-cells, closely mimics not only the phenotype but also the pathogenesis of T2D in humans. Although there are many genetic models that are commonly used as diabetes models, several shortcomings exist with these models in comparison with the HFD/STZ model. On the one hand, overnutrition due to excessive consumption of saturated fat is responsible for the prevalence of T2D. Feeding with a high-fat diet in an animal model is employed to simulate this human situation. Thus, the HFD/STZ model can better simulate the disease progression of diabetes in clinical patients than can spontaneous genetic models. On the other hand, unlike many genetic models that focus on autoimmune processes, induced animal models focus more on hyperglycemia itself and incorporate the development of stable and persistent hyperglycemia. Based on the above discussion, we finally built a T2D model by the HFD/STZ method. Multiple studies have shown that the success rate of this model is approximately 80%, and the mortality rate of this model ranges from 10 to 45% [6, 17], which is consistent with our results. In the present study, 5 rats failed to develop hyperglycemia. We speculated that this may be related to improper injection of STZ into the subdermal space, thereby reducing the success rate. Three rats died of ketoacidosis, and two died of infection. None of these rats died because of diabetic cardiomyopathy. Throughout the experiment, all surviving rats were in good spirits, and no other abnormal manifestations were observed except for the symptoms of obesity, polydipsia, and polyuria. Thus, the T2D model induced by HFD/STZ is suitable for our research purpose, and this model has been adopted as a classic T2D model in many high-quality studies [27].
Diabetic cardiomyopathy is a serious cardiovascular complication of T2D and used to describe myocardial structural and functional changes that occur in patients with diabetes in the absence of coronary artery disease as well as hypertensive, valvular, or congenital heart disorders. Structurally, DCM is characterized by cardiac hypertrophy and myocardial fibrosis. Functionally, DCM initially manifests as isolated diastolic dysfunction, which is characterized by increased E/e’ ratio, inverted E/A ratio, and constant EF and FS, resembling the phenotype of heart failure with preserved ejection fraction. With the development of diabetes, DCM gradually manifests as systolic dysfunction and ultimately progresses to systolic heart failure, resembling the phenotype of heart failure with reduced ejection fraction. In the present study, diabetic rats showed pronounced diastolic dysfunction and preserved systolic function, suggesting that the time point we chose was still in the early stage of DCM.
Diabetes and TMAO
Previous studies have demonstrated that TMAO is closely associated with diabetes. The changes in dietary structure and intestinal flora account for the increased TMAO in diabetic patients [12]. On the one hand, a high-fat diet plays a causal role in the development of insulin resistance and diabetes [12]. In this study, we established a T2D rat model with a long-term high-fat diet, which was rich in choline and hence provided important substrates for TMAO production. Choline provided by the high-fat diet was catabolized into trimethylamine in the intestine, and then trimethylamine was absorbed into the circulation and oxidized to TMAO by hepatic flavin monooxygenase 3 (FMO3). Yoo et al. demonstrated that a high-fat diet could alter intestinal epithelial physiology by impairing mitochondrial bioenergetics in the colonic epithelium and eliminating epithelial hypoxia, ultimately facilitating the production of TMA in the intestine and increasing the levels of TMAO in the circulation [32]. Similarly, a randomized crossover design study also confirmed that a high-saturated-fat dietary pattern could elevate the systemic TMAO level by enhancing dietary precursors and reducing renal TMAO excretion [29]. Consistent with these studies, we also observed a significant increase in plasma TMAO levels in the present study. On the other hand, diabetes changes the composition of the intestinal flora and thus promotes TMAO production [5]. Numerous studies have confirmed that two enzymes encoded by the choline utilization (cut) gene cluster, namely, CutC and CutD, are the key route for converting choline to TMA [18]. Furthermore, Zhu et al. identified that the cut gene cluster was mainly distributed in Firmicutes and Proteobacteria but was conspicuously absent from Bacteroidetes [35]. In our study, we found that the microflora structure of diabetic rats was significantly different from that of control rats. At the phylum level, the relative abundance of Bacteroidetes was significantly decreased, while the relative abundances of Firmicutes and Proteobacteria were increased, especially Proteobacteria. In addition, at the order level, the relative abundances of Clostridiales and Desulfovibrionales were significantly elevated. Specific species among these two orders were verified to harbor choline utilization gene clusters and showed considerable choline consumption [23]. Therefore, these structural changes in the microflora contributed to TMAO generation in diabetic rats. In summary, both dietary structure and gut microbiota are involved in the production of TMAO in diabetes.
TMAO and inflammation
The increased production of TMAO caused by diabetes promoted systemic inflammation. Previous studies have mainly focused on the role of TMAO in the development of vascular inflammation. Seldin et al. reported that both chronic choline feeding and acute TMAO injection were able to induce vascular inflammation by activating mitogen-activated protein kinase and nuclear factor-κB signaling in vitro and in vivo [26]. Similarly, Chen et al. also confirmed that TMAO triggered vascular inflammation both in human umbilical vein endothelial cells and in aortas from ApoE−/− mice, which is associated with the activation of the nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome [3]. Furthermore, numerous clinical studies have confirmed the association between TMAO and systemic inflammation. A cohort study demonstrated that in patients with end-stage renal disease, accumulated TMAO was positively correlated with C-reactive protein levels in serum and IL-6 levels in dialysate, indicating that TMAO changed both the systemic and peritoneal inflammatory status and thus increased the peritonitis risk in patients with chronic kidney disease [34]. Another cross-sectional study showed that patients with elevated TMAO levels had higher plasma concentrations of the inflammatory cytokine TNF-α than healthy participants [22]. All of the above results verified that abnormally increased TMAO promoted systemic inflammation and aggravated inflammatory infiltration in vital organs, such as the heart and kidneys. In the present study, we found that abnormal TMAO in the diabetes group increased the expression of IL-1β, IL-6, and TNF-α in atrial tissues, while DMB treatment alleviated inflammatory cytokine infiltration in atrial tissues by reducing circulating TMAO levels.
TMAO and AF
Atrial inflammation caused by elevated TMAO levels impaired atrial electrical conduction by altering the expression and distribution of gap junction proteins [14]. Gap junctions are clusters of transmembrane channels that mediate electrical coupling and metabolic coupling between adjacent cells. Cx40 and Cx43 are major connexins expressed in the atrial working myocardium [16]. This cell-to-cell communication is crucial for uniform electrical propagation and synchronous contraction. Any changes in connexin expression, distribution, and phosphorylation can lead to heterogeneous repolarization, increased dispersion of refractoriness, and decreased conduction velocity [31]. These abnormalities are conducive to the formation of sustained re-entry and conduction block, ultimately causing the initiation and maintenance of AF. Previous studies have confirmed the effects of inflammation on gap junction dysfunction in the atrium. In vitro, Lazzerini et al. found that IL-6 significantly reduces the protein expression of Cx40 and Cx43 in HL-1 mouse atrial myocytes [16]. In vivo, Sawaya et al. demonstrated that compared with wild-type control mice, transgenic mice with cardiac-restricted overexpression of TNF showed abnormal atrial conduction and increased vulnerability to atrial arrhythmias, which is related to the downregulated expression of Cx40 and disordered distribution of Cx43 [25]. Similarly, in a canine model of sterile pericarditis, altered distributions of Cx40 and Cx43 contributed to markedly abnormal conduction and an increased prevalence of atrial flutter and AF [24]. In our study, we also found that atrial inflammation promoted connexin remodeling, which was characterized by reduced expression of Cx40, decreased phosphorylation of Cx43, and lateralized localization of both connexins. Then, we further detected atrial electrical changes. Previous studies demonstrated that atrial size plays an important role in sustained AF. Larger atrial surface area supports larger number of wavelets, which can propagate simultaneously and reduce the likelihood of fibrillation termination. Thus, AF is easier to generate in larger animals than small animals and small rodents are incapable of sustaining AF [11]. On this basis, we performed burst pacing to induce AF and detected AF vulnerability to reflect atrial electrical changes in the present study. We discovered that the abnormalities of connexin increased the dispersion of the AERP and promoted the susceptibility to AF. DMB treatment ameliorated atrial inflammation and further improved gap junction dysfunction by reducing TMAO levels in the circulation. On this basis, DMB treatment reduced the vulnerability to AF by improving atrial electrical conduction.
Limitations
Several limitations in this study should be acknowledged. Firstly, the electrophysiological studies in the present study were performed in vivo to detect parameters of atrial electrical activity. We did not harvest hearts for Langendorff perfusion in vitro and therefore failed to record the interatrial conduction time. However, the methods we used can better reflect atrial electrical activity under physiological conditions. Secondly, our follow-up time was 3 months. The T2D model at this stage showed obvious intestinal flora dysbiosis and systemic TMAO elevation. Therefore, it is suitable for investigating the effects of TMAO on diabetes-related AF. However, the effects of TMAO on cardiac function and pathological remodeling need to be elucidated with further accumulation of TMAO over time. Thirdly, we mainly detected the pathological changes of the atria. Actually, cardiac inflammation and connexin remodeling induced by diabetes occurred not only in the atria but also in the ventricles. However, the present study aimed to explore the effects of TMAO on atrial electrical changes, so we focused on the changes of atria, and further investigations are required to elucidate the pathological changes of ventricles.
Conclusions
In the development of diabetes, disturbances in the intestinal flora cause the accumulation of TMAO in the circulation. Elevated TMAO levels facilitated atrial inflammation and connexin remodeling, which provided substrates for the initiation and maintenance of AF at the early stage of DCM. Effective reduction of systemic TMAO could alleviate the progression of diabetes-related AF.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bastin M, Andreelli F (2020) The gut microbiota and diabetic cardiomyopathy in humans. Diabetes Metab 46:197–202. https://doi.org/10.1016/j.diabet.2019.10.003
Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA (1994) Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. Jama 271:840–844
Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT (2017) Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc 6. https://doi.org/10.1161/jaha.117.006347
Clemente JC, Manasson J, Scher JU (2018) The role of the gut microbiome in systemic inflammatory disease. BMJ 360:j5145. https://doi.org/10.1136/bmj.j5145
Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E (2016) Diabetes is associated with higher trimethylamine n-oxide plasma levels. Exp Clin Endocrinol Diabetes 124:251–256. https://doi.org/10.1055/s-0035-1569330
Dang JK, Wu Y, Cao H, Meng B, Huang CC, Chen G, Li J, Song XJ, Lian QQ (2014) Establishment of a rat model of type II diabetic neuropathic pain. Pain Med 15(4):637–646. https://doi.org/10.1111/pme.12387_1
Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11:98–107. https://doi.org/10.1038/nri2925
Du X, Ninomiya T, de Galan B, Abadir E, Chalmers J, Pillai A, Woodward M, Cooper M, Harrap S, Hamet P, Poulter N, Lip GY, Patel A (2009) Risks of cardiovascular events and effects of routine blood pressure lowering among patients with type 2 diabetes and atrial fibrillation: results of the ADVANCE study. Eur Heart J 30:1128–1135. https://doi.org/10.1093/eurheartj/ehp055
Foster M, Petocz P, Samman S (2013) Inflammation markers predict zinc transporter gene expression in women with type 2 diabetes mellitus. J Nutr Biochem 24:1655–1661. https://doi.org/10.1016/j.jnutbio.2013.02.006
Fuentes-Antrás J, Ioan AM, Tuñón J, Egido J, Lorenzo O (2014) Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol 2014:847827. https://doi.org/10.1155/2014/847827
Hartley A, Shalhoub J, Ng FS, Krahn AD, Laksman Z, Andrade JG, Deyell MW, Kanagaratnam P, Sikkel MB (2021) Size matters in atrial fibrillation: the underestimated importance of reduction of contiguous electrical mass underlying the effectiveness of catheter ablation. Europace 23(11):1698–1707. https://doi.org/10.1093/europace/euab078
Heianza Y, Sun D, Li X, DiDonato JA, Bray GA, Sacks FM, Qi L (2019) Gut microbiota metabolites, amino acid metabolites and improvements in insulin sensitivity and glucose metabolism: the POUNDS Lost trial. Gut 68:263–270. https://doi.org/10.1136/gutjnl-2018-316155
Huxley RR, Filion KB, Konety S, Alonso A (2011) Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol 108:56–62. https://doi.org/10.1016/j.amjcard.2011.03.004
Hu YF, Chen YJ, Lin YJ, Chen SA (2015) Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol 12:230–243. https://doi.org/10.1038/nrcardio.2015.2
Lazzerini PE, Capecchi PL, Laghi-Pasini F (2017) Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J 38:1717–1727. https://doi.org/10.1093/eurheartj/ehw208
Lazzerini PE, Laghi-Pasini F, Acampa M, Srivastava U, Bertolozzi I, Giabbani B, Finizola F, Vanni F, Dokollari A, Natale M, Cevenini G, Selvi E, Migliacci N et al (2019) systemic inflammation rapidly induces reversible atrial electrical remodeling: the role of interleukin-6-mediated changes in connexin expression. J Am Heart Assoc 8:e011006. https://doi.org/10.1161/jaha.118.011006
Leger T, He B, Azarnoush K, Jouve C, Rigaudiere JP, Joffre F, Bouvier D, Sapin V, Pereira B, Demaison L (2019) Dietary EPA increases rat mortality in diabetes mellitus, a phenomenon which is compensated by green tea extract. Antioxidants (Basel) 8(11):526. https://doi.org/10.3390/antiox8110526
Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP (2015) Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 6. https://doi.org/10.1128/mBio.00042-15
Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, Ramu V, Bhogal S, Kumar G, Shanmugasundaram M, Paul TK (2019) Diabetic cardiomyopathy - a comprehensive updated review. Prog Cardiovasc Dis. 62(4):315–326. https://doi.org/10.1016/j.pcad.2019.03.003
Phang RJ, Ritchie RH, Hausenloy DJ, Lees JG, Lim SY (2022) Cellular interplay between cardiomyocytes and non-myocytes in diabetic cardiomyopathy. Cardiovasc Res cvac049 https://doi.org/10.1093/cvr/cvac049
Plitt A, McGuire DK, Giugliano RP (2017) Atrial fibrillation, type 2 diabetes, and non-vitamin K antagonist oral anticoagulants: a review. JAMA Cardiol 2:442–448. https://doi.org/10.1001/jamacardio.2016.5224
Rohrmann S, Linseisen J, Allenspach M, von Eckardstein A, Müller D (2016) Plasma concentrations of trimethylamine-N-oxide are directly associated with dairy food consumption and low-grade inflammation in a German adult population. J Nutr 146:283–289. https://doi.org/10.3945/jn.115.220103
Romano KA, Vivas EI, Amador-Noguez D, Rey FE (2015) Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 6:e02481. https://doi.org/10.1128/mBio.02481-14
Ryu K, Li L, Khrestian CM, Matsumoto N, Sahadevan J, Ruehr ML, Van Wagoner DR, Efimov IR, Waldo AL (2007) Effects of sterile pericarditis on connexins 40 and 43 in the atria: correlation with abnormal conduction and atrial arrhythmias. Am J Physiol Heart Circ Physiol 293:H1231–H1241. https://doi.org/10.1152/ajpheart.00607.2006
Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS (2007) Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292:H1561–H1567. https://doi.org/10.1152/ajpheart.00285.2006
Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM (2016) Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc 5. https://doi.org/10.1161/jaha.115.002767
Sun Y, Shi H, Yin S, Ji C, Zhang X, Zhang B, Wu P, Shi Y, Mao F, Yan Y, Xu W, Qian H (2018) Human mesenchymal stem cell derived exosomes alleviate type 2 diabetes mellitus by reversing peripheral insulin resistance and relieving β-cell destruction. ACS Nano 12(8):7613–7628. https://doi.org/10.1021/acsnano.7b07643
Wang A, Green JB, Halperin JL, Piccini JP Sr (2019) Atrial fibrillation and diabetes mellitus: JACC review topic of the week. J Am Coll Cardiol 74:1107–1115. https://doi.org/10.1016/j.jacc.2019.07.020
Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, Koeth RA, Li L, Wu Y, Tang WHW, Krauss RM, Hazen SL (2019) Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur Heart J 40:583–594. https://doi.org/10.1093/eurheartj/ehy799
Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE et al (2015) Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163:1585–1595. https://doi.org/10.1016/j.cell.2015.11.055
Wiegerinck RF, van Veen TA, Belterman CN, Schumacher CA, Noorman M, de Bakker JM, Coronel R (2008) Transmural dispersion of refractoriness and conduction velocity is associated with heterogeneously reduced connexin43 in a rabbit model of heart failure. Heart Rhythm 5:1178–1185. https://doi.org/10.1016/j.hrthm.2008.04.026
Yoo W, Zieba JK (2021) High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. 373: 813–8.https://doi.org/10.1126/science.aba3683
Zakkar M, Ascione R, James AF, Angelini GD, Suleiman MS (2015) Inflammation, oxidative stress and postoperative atrial fibrillation in cardiac surgery. Pharmacol Ther 154:13–20. https://doi.org/10.1016/j.pharmthera.2015.06.009
Zhang L, Xie F, Tang H, Zhang X, Hu J, Zhong X, Gong N, Lai Y, Zhou M, Tian J, Zhou Z, Xie L, Hu Z et al (2022) Gut microbial metabolite TMAO increases peritoneal inflammation and peritonitis risk in peritoneal dialysis patients. Transl Res 240:50–63. https://doi.org/10.1016/j.trsl.2021.10.001
Zhu Y, Jameson E, Crosatti M, Schäfer H, Rajakumar K, Bugg TD, Chen Y (2014) Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc Natl Acad Sci U S A 111:4268–4273. https://doi.org/10.1073/pnas.1316569111
Funding
This study was supported by the National Natural Science Foundation of China (No. 81770333, No. 81800350).
Author information
Authors and Affiliations
Contributions
The authors declare that all data were generated in-house and that no paper mill was used. WYJ and JYH designed this study and performed most of the experiments. SCW conducted the rest of the experiments. YTL, YDC, and ZXJ analyzed the data. WYJ and JYH wrote the manuscript. QJS was the corresponding author, and all the experiments were performed under his guidance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All study protocols were approved by the Animal Ethics Committee of Nanjing Medical University (IACUC-2011003) and fully complied with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Keypoints
• Diabetic rats showed disordered gut microbiota and increased TMAO levels.
• Elevated TMAO levels facilitated atrial inflammation and connexin remodeling.
• Elevated TMAO levels increased the susceptibility of diabetic rats to AF.
• DMB alleviated the progression of diabetes-related AF by reducing TMAO levels.
Rights and permissions
About this article

Cite this article
Jiang, WY., Huo, JY., Wang, SC. et al. Trimethylamine N-oxide facilitates the progression of atrial fibrillation in rats with type 2 diabetes by aggravating cardiac inflammation and connexin remodeling. J Physiol Biochem 78, 855–867 (2022). https://doi.org/10.1007/s13105-022-00908-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-022-00908-2