
Abstract
Many studies demonstrated that angiotensin 2 type 1 receptor (AT1R) blockade accelerates renal recovery in post-ischaemic kidney but there are many controversies related to its net effect on kidney structure and function. During the past years, our research group was trying to define the pathophysiological significance of the renin–angiotensin system on post-ischemic acute renal failure (ARF) development in normotensive Wistar as well as hypertensive rats (SHR). This review mostly summarizes our experience in that field. Our previous studies in normotensive rats revealed that AT1R blockade, except slightly renal vascular resistance improvement, had no other obvious beneficial effects, and therefore implies angiotensin 2 (Ang-2) overexpression as non-dominant on kidney reperfusion injuries development. Similarly it was observed in Wistar rats with induced mild (L-NAME, 3 mg/kg b.w.) nitric oxide (NO) deficiency. Expectably, in strong induced (L-NAME, 10 mg/kg b.w.) NO deficiency associated with ARF, massive tubular injuries indicate harmful effects of AT1R blockade, implying strongly disturbed glomerular filtration and suggesting special precaution related to AT1R blockers usage. Opposite to previous, by our opinion, AT1R antagonism promises new advance in treatment of essentially hypertensive subjects who develop ARF. Increased glomerular filtration, diminished oxidative stress, and most importantly improved tubular structure in postishemic SHR treated with AT1R blocker losartan, implicate Ang-2 over production as potently agent in the kidney ischemic injury, partly trough generation of reactive oxygen species. These data contribute understanding the pathogenesis of this devastating illness in hypertensive surroundings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Post-ischemic acute renal failure (ARF) devastates hospitalized normotensive and hypertensive patients inducing their high rate mortality and morbidity. Series of interrelated events including tubular obstruction, passive backflow of filtrate, preglomerular vasoconstriction, and a fall in both glomerular filtration rate (GFR) and renal blood flow (RBF) characterize renal post-ischemic events in humans and animals [11, 15, 24]. Among others, angiotensin 2 (Ang-2) potently affects intrarenal vasoconstriction [7]. It modulates GFR directly by affecting the tone of both efferent and afferent arterioles and also mediates mesangial cell function [17]. Besides, ischaemic renal injury increases plasma renin activity in the initial stages of both clinical and experimental forms of ARF [38]. Ang-2 is one of the main vasoactive signaling molecule involved in the generation of reactive oxygen species (ROS) which are overproduced in the ischemic phase of ARF [27, 9]. It participates in increased expression and activity of one of the major ROS generators, NADPH oxidase [35]. Renal dysfunction caused by overproduction of ROS after ischemia–reperfusion is closely associated with cell membrane peroxidation, mitochondrial dysfunction, inhibition of protein synthesis, DNA damage and inhibition of the antioxidant defense [16, 12]. Also, it is well-known that the increased production of Ang-2 characterizes hypertension [11]. Beyond intrarenal vasoconstriction, high level of Ang-2 harmfully modulates necrotic and apoptotic changes in the kidney tissue during reperfusion period [12, 30]. Many studies demonstrated that Ang-2 type 1 receptor blockade accelerates renal recovery after post-ischaemic kidney injury [27, 18, 19], but there are many controversies related to its net effect on kidney structure and function. During the past years, our research group was trying to define the pathophysiological significance of this important cardiovascular player on ARF development in normotensive as well as hypertensive rats. The present article mostly considers these data.
AT1 blockade in renal ischemia and normal blood pressure
The most effects of Ang-2 are mediated by AT1 receptor including ‘classical’ effects of the renin–angiotensin system (RAS) such as vasoconstriction, renal salt and water retention, central osmo-control and stimulation of cell growth [3]. Kontogiannis and Burns [27] demonstrated that renal ischaemia–reperfusion injury caused an early increase of intrarenal Ang-2 levels, associated with a reduction of mRNA for angiotensinogen and proximal tubular AT1 receptors. They found that blockade of AT1 receptors with losartan accelerated recovery of renal function followed by significant decrease in serum creatinine after bilateral renal pedicle occlusion for 60 min in Sprague–Dawley rats. These authors suggested improved glomerular filtration due to better renal haemodynamics after AT1 receptor blockade treatment. Opposite, in our study [21] renal blood flow non-significantly raised in the group treated with losartan 24 h after 45-min renal ischaemia in Wistar rats. Also, we showed that losartan, except slightly renal artery resistance improvement, did not exert beneficial effects on glomerular filtration rate (Fig. 1a). Finally, in this study losartan had almost no any influence on tubular necrosis induced by ischaemia–reperfusion (Fig. 1b, c). Different findings between our results and Kontogiannis and Burns [27] could be point of both different rat strain and ARF model usage. Namely, Kontogiannis and Burns bilaterally occlude renal pedicle of Sprague–Dawley rats for 60 min, which thereafter recovered both kidneys. These authors used different dose and time course of losartan application (25 mg/kg s.c. starting at the time of reperfusion) compared to our study. Such experimental design was probably characterized with a milder renal injury in comparison to our one kidney 45-min reperfusion model, allowing RAS blockade to express more beneficial on renal tissue. On the other hand, similar to our results, Kim et al. [25] reported that pre-treatment with enalapril and losartan did not prevent reduction of GFR 24 h after ischaemic ARF. These authors used 60-in bilateral obstruction of renal pedicle in rabbits as a model of kidney ischemia–reperfusion injury. They suggested that the late reduction in GFR in post-ischemic kidneys was not mediated by Ang 2, but was mediated, at least in part, by the tubuloglomerular-feedback mechanism. Although these authors use rabbits as a model, it is rationally to conclude that in our design RAS blockade also through decreased arterial and filtration pressure, disturbed glomerular autoregulation and therefore contributed to the slowing down of kidney recovery. It is well-known that blockade of AT1 receptors has a noticeable natriuretic effect. In the study of our group [21], excretion of sodium was markedly higher in losartan-treated rats than in untreated ARF rats 24 h after reperfusion injury (Fig. 1a). Most probably, this increase in urinary sodium excretion in losartan-treated groups was not due to renal haemodynamic deterioration or massive tubular injury but mostly was consequence of AT1 receptors blockade in tubules. Navar [32] demonstrated the presence of angiotensinogen and angiotensinogen mRNA in proximal tubule cells, what indicated that Ang-2 or precursors of Ang-2 were secreted directly into the proximal tubular lumen by the epithelial cells. Micropuncture studies by these authors provided direct evidence that activation of intraluminal AT1 receptors by Ang-2 exerted a substantial stimulatory influence on sodium transport in both proximal and distal tubules. Moreover, RAS axis is dually affected after renal ischemia. Two pathways, angiotensin-converting enzyme (ACE)/Ang2/AT1 receptor and angiotensin-converting enzyme 2 (ACE2)/(Ang-(1–7)/Mas receptor, have opposite effects on renal tissue injury, deleterious and protective, respectively [20, 10]. ACE2 is a modulator of AgII levels and it converts AgII to Ag-(1–7) which binds to Mas receptor in renal tissue antagonizing many deleterious effects of AgII [41]. Barosso et al. [1] have shown that AVE0991, a nonpeptide Mas agonist, attenuated renal functional impairment, decreased the local and systemic inflammatory responses, and reduced glomerular and tubulointerstitial damage in a murine model of AKI induced by bilateral I/R injury. Upstream blockade of RAS axis trough renin inhibition also exerted beneficial effects on renal I/R. Wang et al. [43] shown that pretreatment with renin inhibitor aliskiren, reduced serum creatinine and blood urea nitrogen levels, ameliorated renal histopathological changes, and decreased the apoptosis of cells and leukocyte infiltration in kidney of Sprague–Dawley rats. They also demonstrated that aliskiren ameliorated oxidative stress trough increasing of superoxide dismutase (SOD) and glutathione (GSH-reduced form) levels. Taken together, our results indicate opposite to others, that AT1 receptor blockade has no beneficial neither deleterious effects in normotensive Wistar rats with induced ARF. Discrepancy among our and studies with beneficially effects of RAS blockade in normotensive rats should be considered in contest of strain usage and degree of kidney injury performed.

Kidney biochemistry and histology in normotensive Wistar rats with renal ischemia. a Losartan had no influence on creatinine clearance, but increased renal failure index and fractional excretion of sodium (FeNa+) in Wistar rats with induced acute renal failure. b Wistar rat with induced ARF. Tubular dilatation, intensive interstitial edema and widespread tubular necrosis in cortico-medulary area (PAS × 250). c Wistar rat with induced ARF and treated with losartan (10 mg/kg b.w.). Tubular dilatation, slightly interstitial edema, and tubular necrosis in cortico-medulary area (PAS × 320)
AT1 blockade in renal ischemia with hypertension
Numerous studies indicate that a vasodilatory nitric oxide (NO) has a crucial role in the pathogenesis of ARF, and that spontaneous NO donors may be clinically effective in ischaemic ARF [22, 24, 28]. This motivated our research group to examine effects of both mild and strong NO blockade (induced by acute L-NAME, NG-L-Arginine Methyl ester, treatment) on the course of post-ischemic ARF in Wistar rats. These models mimic NO deficiency in conditions such as some forms of arterial hypertension, heart failure and chronic renal failure. We found losartan beneficial for protein and sodium excretion in the mild hypertensive NO deficiency (induced by 3 mg/kg L-NAME infusion) in Wistar rats [30]. Nevertheless, there were no obvious tubular injury improvements in this group of rats (Fig. 2a, b). On the other hand, in strong NO blockade induced by acute 10 mg/kg L-NAME infusion and followed by severe hypertension, prominent tubular necrosis in the corticomedullary region, tubular dilatation and a huge number of PAS-positive casts in collecting ducts were observed in rats treated with losartan (Fig. 2c, d). Glomerular filtration was significantly reduced in that group of rats. Harmful effects of losartan suggested that vasoconstriction due to strong NO synthesis blockade could not be ameliorated by Ang-2 receptor antagonism which additionally contributed to disturbed glomerular autoregulation.

Kidney histology of Wistar rats with NO deficiency and induced ARF. a Wistar rat with induced ARF and treated with L-NAME (3 mg/kg nw.). Focal necrotic arrays, a huge number of PAS-positive casts in collecting ducts, tubular dilatation with pronounced loss of the brush border of proximal tubular epithelium in the cortex (PAS × 250); b Wistar rat with induced ARF, treated with both L-NAME (3 mg/kg b.w.) and losartan (10 mg/kg b.w.). Slightly pronounced tubular necrosis and pronounced tubular dilatation in the corticomedullary zone (PAS × 250); c Wistar rat with induced ARF treated with L-NAME (10 mg/kg b.w.). Intensive tubular necrosis, tubular dilatation and interstitial edema (PAS × 320); d Wistar rat with induced ARF, treated with both L-NAME (10 mg/kg b.w.) and losartan (10 mg/kg b.w.) and widespread tubular necrosis, tubular dilatation, and a huge number of PAS-positive casts in tubular lumen (PAS × 320)
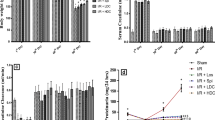
In another set of research, we examined the role of Ang-2 on the course of post-ischemic ARF in spontaneously hypertensive rats (SHR), mainly through relation between the increased oxidative stress and Ang-2 type 1 receptor blockade [18]. We used SHR in which Ang-2-mediated generation of ROS in the kidney is associated with NADPH oxidase overexpression [45], even before the onset of hypertension. It was shown that angiotensin-converting enzyme inhibitor captopril, decreased oxidative stress in hypertensive rats [4]. Additionally, AT1R antagonist, candesartan, significantly attenuated lipid peroxidation in human hypertension [26]. In our study [18], AT1R antagonist losartan reduced TBARS level (a marker of lipid peroxidation) in plasma leading to target cells protection after renal reperfusion injury in SHR (Fig. 3). Increased CAT activity in this study confirmed the influence of Ang-2 on oxidative distress which was reduced by losartan. Authors also showed that losartan could prevent increasing of H2O2 caused by Ang-2 infusion [30]. In previous haemodynamic study [19], we found that losartan, as potent vasodilator, decreased arterial pressure and renal vascular resistance in treated rats compared to control ARF. This suggests that Ang-2 strongly participates in both systemic blood pressure and regional blood flow controls after renal ischemia in SHR. In addition to described haemodynamic benefits, losartan also significantly increased creatinine clearance [18] 24 h after renal reperfusion injury (Fig. 3). These data confirm that in disturbed vasoactive milieu such as hypertension, Ang-2 plays a critical role in control of glomerular filtration. Animal and human based studies found high-density lipoprotein (HDL) fraction of cholesterol protective against ischemia [40, 29]. Thiemermann [40], showed that bolus of HDL could improve renal function and structure after ischemic episode of ARF in Wistar rats. Otherwise, it was shown that Ang-2 downregulated protective HDL receptors on tubular cells via AT1R [44]. In our study [18], blockade of AT1R with losartan resulted in a significant increase of plasma HDL content (that appeared as a main fraction in total plasma), probably due to anti-oxidative action of losartan (Fig. 3). That could be another mechanism of AT1R protection against renal ischemia in hypertensive rats. However, morphological changes in the kidney tissue represent the best portrait of ARF development [21, 37]. Our results [18] clearly indicated that losartan, used to block AT1R in the early stages of ischemic ARF had beneficial effects on renal morphological structure in genetically hypertensive rats. Less severe lesions of tubular epithelial cells (the main damage area during ARF) in losartan-treated rats (Fig. 4a, b) correlated with previously reported improvement of both systemic and renal artery hemodynamic parameters [19], as well as the biochemical markers of kidney function [18]. Other authors also showed protective effect of losartan on kidney morphology in rats with gentamicin-induced ARF [14] and also in malignantly hypertensive rats [39]. Considering cell surviving and regeneration as important factors in recovery after ARF induction, we examined expression of pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) genes in hypertensive rats with ARF [18]. We found that AT1R blockade significantly reduces the expression of both Bax, as well as Bcl-2 genes after kidney reperfusion injury. Because Ang-2-induced apoptosis of mesangial and proximal tubular cells is associated with increased generation of ROS [36], our findings suggest that RAS blockade slows down tubular cell death leading to decreased expression of pro-apoptotic Bax gene after renal ischemia–reperfusion (Fig. 4c, d). On the other hand, decresed oxidative stress and both structural and functional tubular improvement due to AT1R blockade, resulted with diminished anti-apoptotic Bcl-2 gene expression (Fig. 4e, f). In recent years, the relevance of angiotensin type 2 receptors (AT2Rs) in renal ischemia–reperfusion is occupying attention of many research groups. AT2Rs, in many ways negatively modulate the actions of AT1Rs [5, 8, 6, 23], are widely expressed throughout the kidney. In addition to vascular and tubular elements, they are frequently expressed in renal proximal tubule cells [33, 31]. Usage of AT2R transgenic mice subjected to five sixths (5/6) nephrectomy is a relevant model of ischemic renal injury [13]. In those animals, glomerular expression of AT2Rs was upregulated by 5/6 nephrectomy leading to decreasing of urinary albumin excretion in comparison to wild-type mice. Also, transforming growth factor-β and platelet-derived growth factor were significantly reduced in those rats, but urinary excretion of nitric oxide metabolites increased 2.5-fold. All of the above-mentioned responses were blocked by AT2R antagonist PD-123,319 [34]. Moreover, animals pre-treated with AT1R antagonist, losartan, showed a further increase in AT2R expression [42]. These data indicate that AT2R represents a beneficial counter-regulatory mechanism to protect the kidney from the ischemic injury. Opposite to previous, study of Bedford at al. [2] showed that usage of RAS antagonists increased risk of AKI occurring in primary care in all patients even after multiple adjustment for confounding risk factors, importantly including adjustment for systolic blood pressure. Furthermore, these authors showed that after fully adjustments of hypertension the risk fell away and became non-significant for moderate and severe forms of AKI. In summary, our results clearly indicated that AT1R blockade had potential to reduce hypertension and improve kidney structure and function after kidney reperfusion injury in rats with spontaneously hypertension. This also suggests that hypertensive patients on RAS receptor antagonist therapy carry no special risks if suffer episode of post-ischemic AKI. Nevertheless, this conclusion requires serious clinical evaluation.

Biochemical parameters in SHR with renal ischemia. Losartan reduces lipid peroxidation, increases HDL and improves glomerular filtration in SHR with ARF. TBARS (thiobarbituric acid reactive substances) concentration; HDL (high-density lipoprotein) concentration; n—number of animals

Kidney histology, Bcl-2 and Bax expression in SHR with ARF. a SHR with induced ARF. Intensively proximal tubular dilatation and necrosis, massive PAS-positive casts in tubular lumens; b SHR rat with induced ARF and treated with losartan (10 mg/kg b.w.). Moderately tubular necrosis, reduced tubular dilatation and less number of PAS-positive tubular casts; c Bcl-2 expression in SHR with induced ARF. Upregulated Bcl-2 expression on tubular cells; d Bcl-2 expression in SHR with induced ARF and treated with losartan (10 mg/kg b.w.). Slightly expression of Bcl-2 protein on tubular cells; e Bax expression in SHR with induced ARF. Widely and strong expression of Bax expended on proximal tubular cells; f Bax expression in SHR with induced ARF and treated with losartan (10 mg/kg b.w.). Reduced Bax protein tubular expression after ischemic-reperfusion injury in losartan-treated rats
Conclusion
Blockade of the renin–angiotensin cascade remains intriguing point in renal pathophysiology. As an important physiological system that among others regulates glomerular filtration and tubular reabsorption, RAS is over expressed in renal pathology. Nevertheless, its therapeutically blockade requires serial precautions in these conditions. Our studies in normotensive rats revel that AT1R blockade, except slightly renal vascular resistance improvement, had no obvious beneficial effects on kidney function and structure after renal ischemia. This implies Ang-2 overexpression as non-dominant event in reperfusion injuries in Wistar rats. Similarly, it was observed in Wistar rats with mild NO deficiency. Expectably, in the stage with strong NO deficiency, massive tubular injuries and declined glomerular filtration were potentiated with adverse effects of Ang-2 receptor blockade what requires special precaution.
Opposite to previous, AT1R antagonism promises new advance in the treatment of hypertensive subjects who developed ARF. Increased glomerular filtration, diminished oxidative stress, and most importantly improved tubular structure in losartan-treated SHR with induced ARF, implicate that Ang-2 overexpression potently participates in the kidney ischemic injury, partly trough generation of ROS. Further increase of protective AT2R expression after AT1R antagonist therapy, at least in part, could contribute to better function and structure of the kidney. In summary, all presented data participate understanding the pathogenesis of this devastating illness in hypertensive surroundings and for therapy of hypertensive patient who survived ischemic episode of ARF.
References
Barroso LC, Silveira KD, Lima CX, Borges V, Bader M, Rachid M, et al. (2012) Renoprotective effects of AVE0991, a nonpeptide mas receptor agonist, in experimental acute renal injury. Int J Hypertens 2012:808726. doi:10.1155/2012/808726
Bedford M, Farmer CKT, Irving J, Stevens PE (2015) Acute kidney injury: an acceptable risk of treatment with renin-angiotensin system blockade in primary care? Canadian Journal of Kidney Health and Disease 2:14. doi:10.1186/s40697-015-0044-y
Berry C, Touyz R, Dominiczak AF, Webb RC, Johns DG (2001) Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. Am J Phys 281:H2337–H2365
Bolterman RJ, Manriquez MC, Ortiz Ruiz MC, Juncos LA, Romero JC (2005) Effects of captopril on the renin angiotensin system, oxidative stress, and endothelin in normal and hypertensive rats. Hypertension 46:943–947
Carey RM, Siragy HM (2003) Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal disease. Endocr Rev 24:261–271
Carey RM (2005) Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension 45:840–844
Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, et al. (2006) Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A 103:17985–17990
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52:415–472
Fang F, Liu GC, Zhou X, Yang S, Reich HN, et al. (2013) Loss of ACE2 exacerbates murine renal ischemia-reperfusion injury. PLoS One 8:e71433
Ferrario CM (2011) ACE2: more of Ang-(1–7) or less Ang II? Curr Opin Nephrol Hypertens 20:1–6
Finn WF (1980) Enhanced recovery from postischemic acute renal failure. Micropuncture studies in the rat. Circ Res 46:440–448
Gobe G, Zhang XJ, Willgoss DA, Schoch E, Hogg NA, et al. (2000) Relationship between expression of Bcl-2 genes and growth factors in ischemic acute renal failure in the rat. J Am Soc Nephrol 11:454–467
Hashimoto N, Maeshima Y, Satoh M, Odawara M, Sugiyama H, Kashihara N, Matsubara H, Yamasaki Y, Makino H (2004) Overexpression of angiotensin type 2 receptor ameliorates glomerular injury in a mouse remnant kidney model. Am J Physiol Renal Physiol 286:F516–F525
Heeba GH (2011) Angiotensin II receptor blocker, losartan, ameliorates gentamicin-induced oxidative stress and nephrotoxicity in rats. Pharmacology 87:232–240
Honda N, Hishida A (1993) Pathophysiology of experimental nonoliguric acute renal failure. Kidney Int 43:513–521
Inal M, Altinisik M, Bilgin MD (2002) The effect of quercetin on renal ischemia and reperfusion injury in the rat. Cell Biochem Funct 20:291–296
Ishikawa I, Harris RC (1991) Angiotensin actions in the kidney: renewed insights into the old hormone. Kidney Int 4:583–596
Ivanov M, Mihailović-Stanojević N, Grujić Milanović J, Jovović Đ, Marković-Lipkovski J, Ćirović S, Miloradović Z (2014) Losartan improved antioxidant defense, renal function and structure of postischemic hypertensive kidney. PLoS One. doi:10.1371/journal.pone.0096353
Ivanov M, Mihailović-Stanojević N, Grujić Milanović J, Jovović Đ, Miloradović Z (2011) Prevention of systemic and regional haemodynamic alterations, hypercreatininemia, hyperuremia and hyperphosphatemia by losartan in hypertension with acute renal failure. Acta Physiol Hung 98:1–7
Iwai M, Horiuchi M (2009) Devil and angel in the renin–angiotensin system: ACE–angiotensin II–AT1 receptor axis vs. ACE2–angiotensin-(1–7)–mas receptor axis. Hypertens Res 32:533–536
Jerkić M, Miloradović Z, Jovović D, Mihailović-Stanojević N, Elena JV, Nastić-Mirić D, Grujić-Adanja G, Rodriguez-Barbero A, Marković-Lipkovski J, Vojvodić SB, Manero MV, Prieto MP, Lopez-Novoa JM (2004) Relative roles of endothelin-1 and angiotensin II in experimental post-ischaemic acute renal failure. Nephrol Dial Transplant 19:83–94
Jerkić M, Varagić J, Jovović Đ, Radujković-Kuburović G, Nastić-Mirić D, Adanja-Grujić G, Marković-Lipkovski J, Dimitrijević J, Miloradović Z, Vojvodić SB (1999) L-arginine reduces tubular cell injury in acute postischemic renal failure. Nephrol Dial Transplant 14:1398–1407
Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE (2008) AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther 120:292–316
Kakoki M, Hirata Y, Hayakawa H, Suzuki E, Nagata D, Tojo A, Nishimatsu H, Nakanishi N, Hattori Y, Kikuchi K, Nagano T, Omata M (2000) Effects of tetrahydrobiopterin on endothelial dysfunction in rats with ischemic acute renal failure. J Am Soc Nephrol 11:301–309
Kim SJ, Lim YT, Kim BS, et al. (2000) Mechanism of reduced GFR in rabbits with ischemic acute renal failure. Renal Fail 22:129–141
Koh KK, Ahn JY, Han SH, Kim DS, Jin DK, et al. (2003) Pleiotropic effects of angiotensin II receptor blocker in hypertensive patients. J Am Coll Cardiol 42:905–910
Kontogiannis J, Burns KD (1998) Role of AT1 angiotensin II receptors in renal ischemic injury. Am J Phys 274:F79–F90
Matsumura Y, Nishiura M, Deguchi S, Hashimoto N, Ogawa T, Seo R (1998) Protective effect of FK409, a spontaneous nitric oxide releaser, on ischemic acute renal failure in rats. J Pharmacol ExpTher 287:1084–1091
Miller GJ, Miller NE (1975) Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet 1:16–19
Miloradović Z, Jerkić M, Jovović Đ, Mihailović-Stanojević N, Grujić Milanović J, et al. (2007) Bosentan and losartan ameliorate acute renal failure associated with mild but not strong NO blockade. Nephrol Dial Transplant 22:2476–2484
Miyata N, Park F, Li XF, Cowley AW Jr (1999) Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol Renal Physiol 277:F437–F446
Navar LG, Harrison-Bernard LM, Wang CT, Cervenka L, Mitchell KD (1999) Concentrations and actions of intraluminal angiotensin II. J Am Soc Nephrol 10:S189–S195
Ozono R, Wang Z-Q, Moore AF, Inagami T, Siragy HM, Carey RM (1997) Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension 30:1238–1246
Padia SH, Carey RM (2013) AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch 465:99–110
Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, et al. (1996) Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97:1916–1923
Ritz E, Haxsen V (2003) Angiotensin II and oxidative stress: an unholy alliance. J Am Soc Nephrol 14:2985–2987
Silici S, Ekmekcioglu O, Kanbur M, Deniz K (2011) The protective effect of royal jelly against cisplatin-induced renal oxidative stress in rats. World J Urol 29:127–132
Stein HJ, Lifschitz DM, Barnes BL (1978) Current concepts on the pathophysiology of acute renal failure. Am J Phys 234:F171–F181
Therrien F, Lemieux P, Belanger S, Agharazii M, Lebel M, et al. (2009) Protective effects of angiotensin AT1 receptor blockade in malignant hypertension in the rat. Eur J Pharmacol 607:126–134
Thiemermann C, Patel NS, Kvale EO, Cockerill GW, Brown PA, et al. (2003) High density lipoprotein (HDL) reduces renal ischemia/reperfusion injury. J Am Soc Nephrol 14:1833–1843
Tikellis C, Bernardi S, Burns WC (2011) Angiotensin converting enzyme 2 is a key modulator of the renin–angiotensin system in cardiovascular and renal disease. Curr Opin Nephrol Hypertens 20:62–68
Vazquez E, Coronel I, Bautista R, Romo E, Villalon CM, Avila-Casado MC, Soto V, Escalante B (2005) Angiotensin II-dependent induction of AT(2) receptor expression after renal ablation. Am J Physiol Renal Physiol 288:F207–F213
Wang Z, Liu Y, Han Y, Guan W, Kou X, Fu J, et al. (2013) Protective effects of aliskiren on ischemia–reperfusion-induced renal injury in rats. Eur J Pharmacol 718:160–166
Wolf G, Wenzel U, Jablonski K, Brundert M, Rinninger F (2005) Angiotensin II down-regulates the SR-BI HDL receptor in proximal tubular cells. Nephrol Dial Transplant 20:1222–1227
Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, et al. (1998) Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 32:488–495
Acknowledgments
This work was supported by a grant (projects OI175096) from the Ministry of Education, Science and Technological Development of Serbia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Miloradović, Z., Ivanov, M., Jovović, Đ. et al. Angiotensin 2 type 1 receptor blockade different affects postishemic kidney injury in normotensive and hypertensive rats. J Physiol Biochem 72, 813–820 (2016). https://doi.org/10.1007/s13105-016-0514-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-016-0514-4