
Abstract
Sirtuins, silent information regulator 2 (Sir 2) proteins, belong to the family of NAD+-dependent enzymes with deacetylase or mono-ADP-ribosyltransferase activity. These enzymes are responsible for processes of DNA repair or recombination, chromosomal stability and gene transcription. In mammals, sirtuins occur in seven varieties, from 1 to 7 (SIRT1–SIRT7), differing among themselves with location. SIRT1, the best known variety, exerts its effects on proteins via NAD+ coenzymes, being thus associated with cellular energetic metabolism and the ‘red–ox’ state. Its deficits are, among others, concomitant with stressful situations and associated with pathophysiologies of many medical conditions, including diabetes mellitus, cardiovascular diseases, neurodegenerative syndromes and kidney diseases. In kidney disorders, it promotes (stimulates) the survival of cells in an affected kidney by modulating their responses to various stress stimuli, takes part in arterial blood pressure control, protects against cellular apoptosis in renal tubules by catalase induction and triggers autophagy. More and more available in vitro and in vivo data indicate SIRT1 activity to be oriented, among others, towards nephroprotection. Thus, SIRT1 may become a novel element in the therapy of age-related renal diseases, including diabetic nephropathy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Sirtuin family
Sirtuins, i.e. silent information regulator 2 proteins, belong to an old, evolutionary conservative family of nicotinamide adenine dinucleotide (NAD+)-dependent enzymes with deacetylase or mono-ADP-ribosyltransferase [17, 18]. Sirtuins control the processes of DNA repair and recombination, chromosomal stability and gene transcription, and are responsible for health-promoting effects of caloric restriction (CR), including delays of ageing processes in the organism. It is assumed that sirtuins, via evolution, sort of translate the availability of NAD+ (which is a sort of ‘currency’ of the energetic metabolism) into a transcriptive control of the genes, which are important for the metabolic adaptation of cells and of the organism.
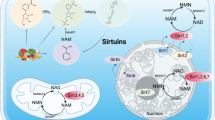
In mammals, 1–7 sirtuins (SIRT1–SIRT7) are observed, differing in their localisation: SIRT1—cell nucleus and cytoplasm; SIRT2—cytoplasm and cell nucleus; SIRT3—mitochondria, cell nucleus and cytoplasm; SIRT4 and SIRT5—mitochondria; and SIRT6 and SIRT7—cell nucleus [17, 18]. A big proportion of SIRT1 is localised in euchromatin, while SIRT6 is found in heterochromatin and SIRT7 is identified mainly in the nucleolus.
Sirtuins have been divided into four classes (SIRT1, 2 and 3 constitute class I; SIRT4 is in class II; SIRT5, class III; SIRT6 and 7 are in class IV); however, there is no relationship between the class assignment of particular sirtuins and their biological function. Sirtuin features have successively transformed to adjust the functions of cells to the ongoing changes in metabolism and environment. Sirtuin activity is mainly related to its post-translational modifications, as well as to the expression of various proteins, including the modulators, and its enzymatic capability.
Metabolic functions of SIRT1 in kidneys
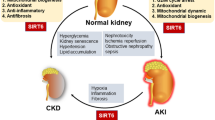
SIRT1 is the most thoroughly studied and evaluated sirtuin. It exerts its effects onto proteins via the NAD+ coenzyme, thus being related with the cellular energetic metabolism and the ‘red–ox’ state. Its deficit in stressful situations (e.g. metabolic and oxidative stress or hypoxia) is associated with pathophysiologies of many diseases, including diabetes mellitus, cardiovascular conditions, neurodegenerative syndromes and renal diseases. See Fig. 1 for an illustration of SIRT1 metabolic functions in the kidney.

Metabolic functions of SIRT 1 in the kidney
Results of in vitro and in vivo studies demonstrate that sirtuins control metabolic pathways in renal cells [8, 17]. SIRT1 promotes the survival of kidney cells by modulating their response to various stress stimuli (oxidative, genotoxic or hypoxic), thus significantly preventing the ageing processes. Any ageing process is inseparably associated with stress-induced increase of apoptosis. SIRT1 activation reduces the process of apoptosis in renal cells of human embryos. In mesangial cells, SIRT1 reduces apoptosis induced by oxidative stress, also protecting them against transforming growth factor beta (TGF-β1)-dependent apoptosis.
The results of studies by Kume et al., published in 2010, demonstrated that the reduced SIRT1 expression rates in aged kidneys of mice was associated with increased oxidative stress and with morphological changes in the mitochondria, such as swelling and disintegration of mitochondrial crests [10]. It has been proven that mitochondrial destruction, as observed in the aged kidneys, could result from Sirt1 deficiency, as Sirt1 enhances cell adaptation to autophagy-stimulating hypoxia. In turn, impaired autophagic flux and deteriorated renal function lead to age-related disability of renal proximal tubular cells in fighting various harmful factors, e.g. drugs or proteinuria.
High SIRT1 concentrations were found in the interstitial cells of the renal medulla, while its low levels were traced in the renal cortex. Some in vitro studies demonstrate protective effects of SIRT1 on the cortex, following hydrogen peroxide administration (protection against oxidative stress). It has been found that a pharmacological induction of SIRT1 exerts a protective effect onto the renal medulla by activating the antioxidative pathways, which increases renal immunity to hypoxia conditions or to oncotic pressure changes [4, 7].
Beside SIRT1 levels, it is, among others, NAD+ concentration that is of fairly high significance. According to Hao and Haase, the NAD+ salvage pathway plays a critical role in the regulation of SIRT1 enzymatic activity by lowering the concentration of inhibitory NAM and by increasing levels of sirtuin co-substrate NAD+ [4].
He et al. found large quantities of SIRT1 in the interstitial cells of murine renal medulla, where it probably increases the resistance to oxidative stress [7]. It is known that renal medulla is under a high risk of oxidative stress because of particular local hypoxia and hypertonicity.
In mice deprived of the SIRT1 gene, increased quantities of apoptosis and fibrosis were found following an experimental kidney injury (unilateral occlusion of the ureter). SIRT1 activation, for example by an administration of resveratrol (3,5,4-trihydroxystilbene), improves the survival of cells and reduces the number of apoptoses and the degree of fibrosis in response to oxidative stress. Additionally, SIRT1 deficit decreases the level of induced COX2 in the parenchyma of renal medulla under oxidative stress (exogenous PGE2 decreases apoptosis in oxidative stress in SRT1-deprived cells), which proves COX2 to be one of the mediators of SIRT1 activity [7]. The authors proposed that Sirt1 should be treated as a therapeutic goal for reducing the possible damage of kidney medullary cells resulting from oxidative stress [7].
The TGF-β1–Smad3 signal pathway plays a key role in the pathogenesis of kidney fibrosis process, which accompanies renal diseases. TGF-β1 activation, both in kidney fibroblasts and tubules epithelium, triggers Smad3 acetylation and phosphorylation.
Li et al. have demonstrated that SIRT1 activation with resveratrol inhibits Smad3 acetylation, resulting in reduced expression of collagen IV and fibronectin induced by TGF-β1 in model animals as well as in tissue cultures of rat fibroblasts [12]. In opinion of the quoted authors, the process of Smad3 deacetylation could be perceived as a new therapeutic means to treat fibrotic diseases [12].
In experiments of Hasegawa et al., they found out that SIRT1 protected the cells in the renal tubules against apoptosis by inducing catalase (via deacetylation of FOXO3) in culture of cells from proximal renal tubules [5, 6].
SIRT1 induces autophagy by deacetylating various factors, such as Atg5, Atg7 and LC3, contributing to the adaptation of cells to stress conditions, thus to their survival [11, 16]. SIRT1 reduces the number of oxidative stress-induced apoptotic processes in mesangial cells by suppressing the activity of p53 protein and attenuates the signalling pathway induced by TGF-β. Therefore, it is assumed that SIRT1 may, in some future perspective, be used as a pharmacotherapy to treat diabetic nephropathy [8, 17].
Some studies have demonstrated that SIRT1-level reduction leads to an increased inflammatory response in adipocytes, monocytes/macrophages and endothelial cells. SIRT1 probably acts as a negative regulator of the nuclear factor kappa B (NF-κB) pathway by Lys310 deacetylation of p65 protein [4, 8]. In their article, Kitada et al. reported anti-inflammatory effects of caloric restriction, noting decreased levels of SIRT1 and elevated levels of acetylated NF-κB in Wistar fatty (fa/fa) rats and increase in SIRT1 levels followed by higher percentage of deacetylated NF-κB after caloric restriction [9].
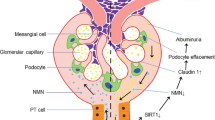
Some presence of SIRT1 was also identified in podocytes, but, thus far, its role in influence on podocyte metabolism has not yet been studied [8]. SIRT1 modulates the cellular response to hypoxia via an interaction with the α subunit of the HIF-2 factor (an oxygen sensitive transcriptive factor). It is then associated with the induction of erythropoietin under hypoxia (and the hypoxia-associated induction of the endothelial growth factor of vessels and of other oxygen-associated genes) [4, 13]. The deacetylating goals, which mediate this task, include, among others, FOXO, p53, thermal shock proteins 1, NF-κb, Ku70 and Smad7 [4, 13].
A reduction of SIRT1 level may lead to impaired reaction to hypoxia via an inactivation of HIF-2a, resulting in chronic renal failure [7, 8]. Studies on the role of SIRT1 suggest also its participation in arterial blood pressure control. This control probably occurs on two planes: vascular tension control and renal control of atrium reabsorption. The observations of Miyazaki et al. have demonstrated that SIRT1 overexpression reduces the expression of AT1 receptor of angiotensin II (AT1R) in cells of vascular smooth muscles, while the inhibitor of SIRT1 (nicotinamide-NAM) increases this expression [15]. The quoted Japanese investigators were the pioneers in showing a downregulation of vascular AT1R by resveratrol [15]. Their published results revealed that resveratrol suppressed AT1R gene expression at the transcriptional level rather than at the post-transcriptional level. The authors stated that resveratrol may suppress renin–angiotensin system not via ACE downregulation but via AT1R downregulation and inhibition of AT1R signalling [15].
It was found out by Miyazaki et al. that an over-expression of SIRT1 inhibits AT1R expression and suppresses angiotensin II-induced ERK phosphorylation; thus, a downregulation of AT1R may attenuate AT1R signalling [15]. In turn, nicotinamide, a natural inhibitor of SIRT1, turned out to increase AT1R expression and, simultaneously, to upregulate the expression level of AT1R mRNA; the overexpression of SIRT1 proved to significantly suppress AT1R protein expression. It was also documented that resveratrol downregulated vascular AT1R by reducing AT1R promoter activity without influencing mRNA stability. Furthermore, resveratrol-induced suppression of AT1R was found to be reversed by nicotinamide. In conclusion, the quoted authors have stated that ‘resveratrol suppresses AT1R expression through SIRT1 activation at least in part’ [15].
Deacetylating the endothelial synthesis of nitrogen oxide, SIRT1 increases the endothelial concentrations of NO, thus promoting vasodilatation [7, 8]. In the kidney, SIRT1 participates in sodium metabolism control, suppressing the transcription of α subunit of the endothelial natrium channel (ENaC). It has been confirmed in some in vitro studies on medullary cells of collecting tubules. Such an activity, rather untypical for sirtuins, is independent of the deacetylating function of SIRT1. It occurs via an interaction with Dot1–methyltransferase, methylating the medullary domain of histone H3 (SIRT1/Dot1 interaction), leading to hypermethylation of chromatin and suppressed expression of the ENaC alpha subunit. This control is also independent of mineralocorticoid signalling [4].
Clinical observations and studies on animals have demonstrated that SIRT1 controls lipid metabolism in the kidneys by inducing LXR (cholesterol level control) and FXR, and inhibiting SREBP, thus suppressing progression of kidney disease [8]. A large group of non-histone proteins serve for a substrate for SIRT1, including transcriptive factors and transcriptive co-regulatory proteins. The influence of NAD+ on deacetylation reactions is considered to bind the deacetylating activity of sirtuins with cell metabolism.
SIRT1 controls energetic metabolism, thus mediating the life-span increasing effect of CR, by promoting gluconeogenesis and suppressing glycolysis in the liver. McCay et al. reported in 1935, and for the first time at all, that rats (submitted to CR) revealed longer, mean life-span values [14], while in laboratory mice (deprived of SIRT1 gene), degenerative changes were observed much earlier (already after 12 months) and CR had no protective value at all [1]. In subsequent years, numerous studies demonstrated that CR delays the ageing process in various species (fungi, worms, flies and rats). Colman et al. also observed CR to have delayed the occurrence of age-related conditions, including diabetes mellitus, cancer, cardiovascular diseases and atrophy of brain neurons, decreasing the mortality rates among apes [3]. Cohen et al. assume that a chronic limitation of calorie supply increases SIRT1 levels [2]. They have stated that a process of SITR1 induction via caloric restriction is an ‘evolutionarily ancient biological stress response that slows ageing by increasing the long-term function and survival of critical cell types’ [2].
It has also been demonstrated that SIRT1 increases the sensitivity to insulin via the modulation of the insulin-related information cascade. Additionally, SIRT1 may control phosphorylation of tyrosine of insulin-induced substrate of the insulin receptor 2 by affecting its acetylation. SIRT1 activity is also correlated with insulin secretion. SIRT1 overexpression in B cells of the pancreatic islets increases the production of ATP by suppressing the expression of the mitochondrial UCP2, thus leading to closure of the ATP-sensitive potassium channels and, in consequence, to insulin secretion. This is why such a big role is assigned to SIRT1 in the pathogenesis of diabetic nephropathy [4].
Numerous scientific observations demonstrate SIRT1 expression in the kidney, as well as its function as a renal survival stimulating factor, although is mediatory character in the CR renoprotective effects has not yet been fully unveiled. Studies on age-related nephropathy and diabetic nephropathy models indicate less renal lesions after CR, probably in result of its systemic and local (tissue-specific) activity, including improved glucose and lipid metabolism profiles, reduced accumulation of advanced glycosylation products, reduced oxidative stress, improved nitrogen oxide balance and lower activity of angiotensin II.
Summing up, it should be emphasised that in pathophysiology of systemic ageing, including the kidneys, a significant role is assigned to factors that influence the signalling pathways associated with cellular energetic metabolism and the ‘red–ox’ state. The recent years of studies on SIRT1 have confirmed its beneficial effects on oxidative stress-induced immunity of cells, reduction of fibrosis, suppression of apoptosis and inflammatory process, induction of autophagy and arterial blood pressure control. The general effect of these processes is nephroprotection; thus, SIRT1 has all the perspectives to be considered a novel therapeutic element in age-related kidney diseases, including diabetic nephropathy.
Abbreviations
- ACE:
-
Angiotensin-converting-enzyme
- Atg5:
-
Autophagy protein 5
- Atg7:
-
Autophagy protein 7
- ATP:
-
Adenosine-5'-triphosphate
- COX2 :
-
Cyclooxygenase-II enzyme
- CR:
-
Caloric restriction
- DNA:
-
Deoxyribonucleic acid
- Dot1:
-
Disruptor of telomeric silencing-1
- ENaC:
-
Epithelial sodium channel
- ERK:
-
Extracellular signal-regulated kinases
- FOXO3 :
-
Forkhead box O3
- FXR:
-
farnesoid X receptor
- HIF-2a:
-
hypoxia-inducible factor
- Ku70:
-
Protein that, in humans, is encoded by the XRCC6 gene
- LC3:
-
protein light chain 3
- LXR:
-
liver X receptor
- Lys310:
-
lysine 310
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NAM:
-
Generating nicotinamide
- NF-κB:
-
Nuclear factor kappa B
- NO:
-
Nitrogen oxide
- PGE2 :
-
prostaglandin E2
- AT1R:
-
angiotensin II receptor, type 1
- SIRT:
-
Silent information regulator
- Smad3:
-
Mothers against decapentaplegic homolog 3
- SREBP:
-
sterol regulatory element binding proteins
- TGF-β1:
-
Transforming growth factor beta
- UCP2:
-
uncoupling protein 2
References
Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J (2007) SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 6(6):759–767
Cohen HY, Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305(5682):390–392
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasle TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R (2009) Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325(5937):201–204
Hao CH, Haase VH (2010) Sirtuins and their relevance to the kidney. J Am Soc Nephrol 21(10):1620–1627
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Washida N, Tokuyama H, Hayashi K, Itoh H (2008) Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372(1):51–56
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M (2010) Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem 285(17):13045–13056
He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC (2010) Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest 120(4):1056–1068
Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D (2013) Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci 124(3):153–164
Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D (2011) Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res 2011:908185
Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, Koya D (2010) Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120(4):1043–1055
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 105(9):3374–3379
Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ (2010) Resveratrol inhibits renal fibrosis in the obstructed kidney: potential role in deacetylation of Smad3. Am J Pathol 177(3):1065–1071
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol Cell 38(6):864–878
McCay CM, Crowell MF, Maynard LA (1989) The effect of retarded growth upon the length of life span and upon the ultimate body size. J Nutr 5(3):155–171
Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K (2008) SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 28(7):1263–1269
Stępień A, Izdebska M, Grzanka A (2007) The types of cell death. Postepy Hig Med Dosw 61(9):420–428
Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin functions in health and disease. Mol Endocrinol 21(8):1745–1755
Yu J, Auwerx J (2009) The role of sirtuins in the control of metabolic homeostasis integrative physiology. Ann NY Acad Sci 1173(1):E10–E19
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Polak-Jonkisz, D., Laszki-Szcząchor, K., Rehan, L. et al. Nephroprotective action of sirtuin 1 (SIRT1). J Physiol Biochem 69, 957–961 (2013). https://doi.org/10.1007/s13105-013-0268-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-013-0268-1