
Abstract
The aim of the study was to analyze the phenotypic and epigenetic changes induced by the shift to a chow diet after an obesogenic environment. Animals were randomized to fed chow (control group) or high-fat–sucrose diet (HFS). After 10 weeks, half of the rats fed with HFS diet were reassigned to a chow diet (rest group) while the other half continued with the obesogenic diet (HFS group) until week 20. Changes in fat content, biochemical profile, and DNA methylation levels of several gene promoters from retroperitoneal adipocytes were analyzed. HFS diet intake for 10 weeks induced obese phenotype in the animals, increasing body weight and fat content. These effects were maintained until the end of the trial in HFS group, where an increase in liver fat content, a modification of lipid profile, and retroperitoneal adipose tissue hypertrophy were also observed. Changing the dietary pattern reversed these parameters. Epigenetic analysis showed that HFS diet intake for 20 weeks hypermethylated several CpG sites (6.7 and 29.30) and hypomethylated CpG site 15 from leptin gene promoter. Moreover, the obesogenic diet also hypomethylated CpG site 1 from Fasn (fatty acid synthase) gene promoter, without changes on Ppargc1a (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), Srebf1 (sterol regulatory element-binding transcription factor 1), and aquaporin 7. Shifting to a chow diet reverted HFS-induced DNA methylation levels of some CpG sites of leptin promoter. Changing the dietary pattern hypomethylated a CpG site of Srebf1 and hypermethylated other CpGs on Ppargc1a and Fasn promoter. This study shed light on the reversibility of phenotypical and epigenetic changes induced by a HFS diet intake.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity has been described as a clinical condition that may have adverse effects on health, leading to reduced life expectancy and/or increased health complications [11], and it has become one of the most serious health problems of the modern society [40]. The excessive accumulation of adipose tissue that characterises obesity is considered an important risk factor for the development of diseases such as type 2 diabetes and cardiovascular disease [14]. The rise in the prevalence of obesity appears to be the result of changes in food intake, eating habits, and physical activity levels [34].
Overweight and obese individuals who lose at least 10 % of their body weight can reduce their risk to suffer cardiovascular disease [2, 43] and type 2 diabetes [26] and can increase the quality of life [17]. Restriction of energy intake is the most commonly used strategy for weight loss in human [30]; however, rodent and human subjects have shown, during energy restriction, sustained reductions in energy expenditure, leading to weight gain when food is available post-restriction [22, 52].
The epigenetic marks are particularly prone to change during embryogenesis [41], gestation, and lactation [35]. However, it has been demonstrated that some environmental changes could also induce variations in DNA methylation profile during adult life [45], representing an excellent target to understand the impact of the environment on the physiological functions. Thus, epigenetic regulation of gene expression has emerged in the recent years as one of the factors involved in the interindividual differences found in different pathologies such as obesity [4]. Bouchard et al. [8] had previously described that the weight loss induced by a hypocaloric diet could alter the DNA methylation profile of specific regions containing genes known to be involved in weight control in subcutaneous adipose tissue. Moreover, it has been reported that baseline DNA methylation patterns of obese individuals could also be helpful to predict the response to a low-caloric diet [15, 36].
In rodents, the impact of the dietary pattern on epigenome has been also studied. Thus, our research group has previously reported changes in the DNA methylation profile of the promoter region of several genes in hypothalamus [45], white adipose tissue, and liver. Specifically, changes in DNA methylation levels due to a high-fat diet intake was found in the promoter region of leptin in retroperitoneal adipose tissue [37] and in the promoter region of fatty acid synthase in the liver [16, 32].
Since the diet is one the most determinant modifiable environmental factors and the dietary pattern has been described to affect the epigenome [44], the aim of this study was to investigate whether the shift to a chow diet reverts the alterations induced by an obesogenic environment, changing not only the phenotypical features but also the DNA methylation profile of the promoters of obesity-related genes.
Material and methods
Animals and experimental design
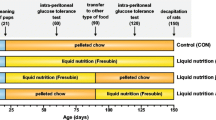
Thirty-six 3-week-old male Wistar rats supplied by Harlan Iberica (Barcelona, Spain) were housed in a temperature- (21 ± 1 °C) and humidity-controlled (55 ± 5 %) room with food and water freely available on a 12-h light cycle (from 8 am to 8 pm). The animals were randomized by weight into two different dietary groups for 10 weeks: chow diet (16.6 % protein, 10.3 % fat, 73.1 % carbohydrate, and 6.5 % sugar from Harlan Iberica, Barcelona, Spain (2014 Teklad Global 14 % Protein Rodent Maintenance Diet)) (n = 12) and high-fat–sucrose (HFS) diet (20 % protein, 45 % fat, and 35 % carbohydrate, from which 23.1 % was sucrose from Research Diets, Inc. (D12451)) (n = 24). After the 10 weeks, 12 animals from the HFS dietary group were separated and fed a chow diet for 10 weeks (Rest group), while the rest of the rats maintained the same diet during this period (control group and HFS group). Animals had ad libitum access to water and food during the dietary treatment (20 weeks), and body weight was recorded weekly.
Body composition
Magnetic resonance technology was used to estimate in vivo fat and lean mass using an Echo MRI Analyzer system (Echo Medical Systems, Houston, TX, USA) [42]. Body composition was measured at several points of the experiment: before starting the dietary treatment (week 0), at weeks 6 and 9 (before the dietary shift), week 17, and at the end of the trial before sacrificing the animals (week 20).
Obtaining serum and tissues
At the end of the experiment, the animals were killed; trunk blood was collected, and serum was stored at −20 °C for further analysis. Liver and retroperitoneal adipose tissues were carefully dissected, weighed, and stored at −80 °C until analyzed. All the procedures performed agreed with the National and Institutional Guidelines of the Animal Care and Use Committee at the University of Navarra.
Liver fat content analysis
Liver was isolated from the animal, and one of the lobes was dissected (always the same), weighted, and stored immediately in appropriately labelled foil at −80 °C for further analysis. Echo MRI Analyzer system (Echo Medical Systems, Houston, TX, USA) [42] was used to estimate the liver fat content.
Serum measurements
Total cholesterol (Cholesterol CP; ABX diagnostic, Montpellier, France), high-density lipoprotein (HDL) cholesterol (HDL direct CP; ABX diagnostic, Montpellier, France), and triglycerides (Triglycerides; Randox Laboratories Ltd., Crumlin, UK) were measured in automatized PENTRA C200 system (HORIBA Medical, Montpellier, France). Low-density lipoprotein (LDL) cholesterol was calculated according to the following formula: Total cholesterol (milligrams per deciliter) − HDL-c (milligrams per deciliter) − (triglycerides (milligrams per deciliter)/5) [18].
Adipocytes isolation
After collecting the adipose tissue, retroperitoneal white adipose tissue samples were cut into small pieces (2 g), and the fragments were digested at 37 °C with collagenase I (2.5 mg/mL) (Worthington Biochemical, Lakewood, NJ, USA) in Krebs–Ringer bicarbonate buffer (KRBA) containing albumin (3.5 g/100 mL) and glucose (6 mM) at 7.4 pH. The ratio of digestion solution for the adipose tissue mass was 2 mL/g. After 30 min of incubation under continuous vigorous shaking (60 cycles/min), the fat cells were filtered through a nylon mesh (400 μm) and washed three times with KRBA to eliminate the stroma–vascular fraction and collagenase. Adipocytes were immediately frozen at −80 °C for further analysis.
DNA methylation analysis
DNA was isolated from retroperitoneal adipocytes using the QiamP DNA Mini KIT (Qiagen, Germantown, MD, USA), and its quality was assessed with PicoGreen dsDNA Quantitation Reagent (Invitrogen, Carlsbad, CA, USA). Genomic DNA was treated with bisulfite by using the EpiTect Bisulfite Kit (Qiagen, Valencia, CA, USA), and DNA methylation, mainly of cytosine nucleotide at the carbon 5 position which is often found in the sequence context cytosine followed by a guanine (CpG), was measured by SEQUENOM MALDI-TOF mass spectrometry [45]. For this purpose, primer pairs were designed to generate amplicons (300–500 bp) covering the relevant CpG sites (Table 1).
Histological analysis
Small pieces of retroperitoneal fat depot were kept in formaldehyde for 24 h and then in ethanol for histological analyses. These tissues were fixed, sectioned at 4 microns, stained with hematoxylin/eosin, and then imaged at ×20 using an Axio Imager M1 Zeiss microscope. The acquired images were stored in uncompressed 24-bit color TIFF format. These images were analyzed with Adiposoft software in order to determine adipocyte diameters and adipose tissue cellularity [7, 19]. Adipocyte number in each tissue was estimated according to formulas from Lemonnier [28].
Statistical analysis
All results are expressed as mean ± standard error the mean (SEM). Statistical analyses were performed using Student’s t test for data before shifting and one-way analysis of variance (ANOVA) and DMS post hoc test after shifting. The level of probability was set at p < 0.05 as statistically significant. All analyses were performed using SPSS 15.0 packages of Windows (Chicago, IL, USA).
Results
Body measurements and food intake
During the first nutritional period (10 weeks), the HFS diet intake induced the expected overweight model, which was reflected by the increase of body fat content (Fig. 1) and body weight (Table 2). Shifting to a chow diet decreased the total body fat content of the animals (Fig. 1), although no significant differences were found in total body weight when comparing HFS group against rest group (Table 2). When body weight gain after dietary change was calculated (differences between week 20 and week 10), HFS group gained significantly more weight than control and rest groups. Furthermore, the animals subjected to HFS diet had a higher energy intake (Table 2) during both nutritional periods, being calorie intake reduced to control group values by the shifting to a chow diet.

Body fat content. Data are mean ± SEM. Statistical analyses were performed using Student’s t test for data before shifting and one-way ANOVA test and DMS post hoc test after shifting. **p < 0.01 and ***p < 0.001. Different letters (a, b) indicate significant differences between groups of at least p < 0.05. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet
HFS diet intake significantly increased hepatic fat content (percent) when compared with control group (Table 2). Interestingly, rats that shifted from a HFS diet to the control diet had similar amount of hepatic fat content than control group.
Biochemical measurements
Regarding the biochemical profile, although there were no significant differences in total cholesterol levels among the groups, HFS diet intake during 20 weeks decreased serum levels of HDL-cholesterol, triglycerides, and free fatty acids in the rats (Table 2). In addition, the animals fed this obesogenic diet showed increased LDL-cholesterol and homocysteine levels. Shifting to a chow diet from weeks 10 to 20 reversed all these changes, except for LDL-cholesterol values (Table 2).
Histological analysis
Results from histological studies showed that HFS intake induced adipose tissue hypertrophy, reflected in a lower number of adipocytes per gram of tissue (Fig. 2b), rather than changes in cell number. Shifting to a chow diet markedly reversed the enlargement of adipocyte volume induced by HFS diet intake increasing the number of adipocytes per gram in rest group animals (Fig. 2b).

Cellularity of retroperitoneal adipose tissue at the end of the trial. a Adipocytes volume and b adipocytes density. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and DMS post hoc test. Different letters (a, b) indicate significant differences between groups of at least p < 0.05. c Three representative images from hematoxylin and eosin-stained paraffin sections of retroperitoneal fat pad. d Adipocyte frequency distribution in retroperitoneal adipose tissue. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet
DNA methylation levels
An epigenetic study was conducted in isolated retroperitoneal adipocytes determining changes in DNA methylation pattern on the promoters of several selected genes (Srebf1, Leptin, Aquaporin-7 (Aqp7), fatty acid synthase (Fasn), and Ppargc1), which have been described as relevant in diet-induced obesity models in rats (Table 1 and Supplementary data).
First, when compared with the control group, we found that HFS diet intake for 20 weeks induced a hypermethylation on leptin promoter in CpG sites 6–7 and 29–30 and a hypomethylation on CpG site 15 (Fig. 3a). We also observed significant changes in DNA methylation levels of two CpG sites of peroxisome proliferator-activated receptor gamma coactivator 1 (Ppargc1a) promoter, resulting in a hypomethylation of CpG sites 3 and 5 (Fig. 3a). Moreover, a hypomethylation induced by HFS exposure was found in CpG sites 1 and 5 of Fasn promoter (Fig. 3b). Srebf1 and Aqp7 promoters did not show statistical changes in DNA methylation due to HFS diet intake (Figure 3b and Supplementary Figure 5b).

DNA methylation levels of individual CpG sites of obesity-related gene promoters. a High methylated promoters and b low methylated promoters. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and DMS post hoc test. Different letters (a, b, c) indicate significant differences between groups of at least p < 0.05. t indicates a trend to significance. Ppargc1a, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Fasn, fatty acid synthase; Srebf1, sterol regulatory element-binding transcription factor 1; HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet
In order to achieve a deeper understanding of the reversion capability of epigenetic marks induced by HFS diet intake, a shift to chow diet was conducted in rats. In this trial, a significant reversion of DNA methylation was observed in CpG sites 29–30 of leptin promoter (Fig. 3a). In addition, CpG sites 6–7 of leptin and CpG sites 3 and 5 of Ppargc1 promoter show a similar pattern, although without statistical significant differences (Fig. 3a). On the other hand, methylation analysis due solely to the shift to a chow diet in DNA from isolated retroperitoneal adipocytes showed a hypomethylation of Srebf1 promoter (CpG site 23) (Fig. 3b). Moreover, CpG sites 15–16 were also hypermethylated due to the shift to a chow diet in Fasn promoter (Fig. 3b). Finally, no differences in methylation levels of any of the analyzed CpG sites of Aqp7 gene promoter were either found by the shift to a chow diet (Supplementary Figure 5b).
Discussion
The main purpose of this study was to determine the potential reversibility of the epigenetic modifications induced by an obesogenic diet (HFS diet) by reducing body weight and adiposity due to the intake of a normocaloric chow diet.
It has been repeatedly described that HFS diets induce an increase of body weight and adiposity in Wistar rats that is usually accompanied by different obesity-related complications such as liver steatosis and insulin resistance [6, 45]. However, in opposition to the hyperlipidemia often found in human diet-induced obesity, rats fed the HFS diet show a decrease of serum triglycerides, HDL-cholesterol, and free fatty acids [46]. As in other studies, histological analyses show that the intake of a HFS diet induces the hypertrophy of retroperitoneal adipocytes [7]. In the last years, several studies have shown that diet-induced obesity is accompanied, in Wistar rats, by alterations in the DNA methylation pattern of important metabolic genes in different tissues, such as leptin in retroperitoneal adipocytes [37], Fasn and NADH dehydrogenase (ubiquinone) 1β-subcomplex 6 (NDUFB6) in epididymal adipose tissue [32], glucokinase, and L-type pyruvate kinase in liver [24], melanocortin-4 receptor (MC4R) in brain [53], or dopamine active transporter (SLC6A3) in hypothalamus [45]. These epigenetic modifications, which could be attributed directly to the hypercaloric diet or to the development of obesity and insulin resistance, might involve changes in gene expression that could impair metabolic or neurological functions [12].
As reported in similar animal models of obesity and metabolic syndrome [39], the normocaloric chow diet exerted an anti-adipogenic/anti-diabetogenic effect and even alleviated liver steatosis, which appears to be due to its lower caloric density and/or its higher fiber content. The shift to chow diet also improved the HFS diet-induced dyslipidemia, normalizing serum HDL, triglycerides, free fatty acids, and homocysteine levels.
The hypertrophy observed in the adipocytes from HFS group was markedly reversed by the shift to a chow diet, observing no differences in both adipocyte size and number between control and rest group. In this sense, the source and amount of energy can modify the adipose tissue growth inducing hypertrophy and hyperplasia and originate metabolic pathologies and adipose tissue dysfunction [7]. In this sense, DNA methylation may contribute to the determination of cells of adipogenic fate and may play a role during adipocyte terminal differentiation, being involved in regional differences in white adipose tissue gene expression and function [47]. In fact, DNA methylation profile is specified by a complex interplay between environmental factors (i.e., the diet) and genetic factors, subject to tissue-specific influence [21].
Apart from obesogenic hypercaloric diets, the epigenetic pattern may respond to different dietary treatments that have been proposed as putative therapeutic agents in the treatment of different metabolic syndrome traits, including methyl donors (vitamins B12, B6 and folic acid, betaine, choline, and methionine), which could prevent the development of nonalcoholic fatty liver, and different polyphenols such as genistein or tea catechins [38] that could protect from obesity by modifying the epigenome. Other studies have demonstrated that weight loss due to hypocaloric diets alter the methylation pattern of different genes in adipose tissue [8] and in peripheral blood mononuclear cells [36]. But the hypothesis of the current works tries to demonstrate that, with only a shift from a hypercaloric to a normocaloric diet, some of the obesogenic diet-induced alterations in the DNA methylation pattern could be reversed in adipocytes of visceral origin. In this study, methylation analysis of DNA from isolated adipocytes was performed, analyzing several gene promoters related to obesity such as leptin, Srebf1, Ppargc1a, Fasn, and aqp7.
Leptin is an adipokine with key functions in the regulation of body weight, by modulating both appetite and energy expenditure, and in which promoter methylation is modulated by different factors such as high-fat diet [37], gestational impaired glucose tolerance [9], and perinatal undernutrition [25]. In this study, a hypermethylation in CpG sites 6–7 and 29–30 and a hypomethylation of CpG site 15 of leptin gene promoter were found due to the intake of HFS diet for 20 weeks. Interestingly, the shift to a chow diet after an exposure to an obesogenic diet for 10 weeks reverted these results, particularly in CpG sites 29–30. These results reinforce the relevance of the study of leptin promoter DNA methylation in adipose tissue and underlie an epigenetic mechanism regulating the dynamics of the expression of leptin.
The Ppargc1a is a co-activator of nuclear receptors and other transcriptions factors that is involved in mitochondrial biogenesis, fatty acid oxidation, insulin-regulated gluconeogenesis, and glucose uptake in peripheral tissues [50]. It provides a direct link between external physiological stimuli and the control of blood pressure, cellular cholesterol homoeostasis, glucose-induced secretion of insulin by B-cells, and obesity development [29]. Different studies have evidenced an epigenetic control of Ppargc1a gene expression. For instance, Ppargc1a gene promoter methylation showed a twofold increase in human diabetic islets compared with non-diabetic islets [29]. Other factors that influence methylation levels of Ppargc1a promoter, at least in human muscle, are high-fat feeding [10, 23] and physical inactivity [1], whereas an association between insulin resistance and liver Ppargc1a DNA methylation has been found in patients with nonalcoholic fatty liver disease [49]. This is the first study that examines the influence of a hypercaloric diet on the methylation pattern of Ppargc1a promoter in adipocytes, but we did not find significant differences. However, the shift to a chow diet increased the methylation levels of two CpG sites (3 and 5). As Ppargc1a mRNA levels are lower in adipose tissue of rats fed a chow diet than in high-fat-fed animals [3], it is reasonable to think that the lower expression of this gene is associated to higher methylation levels of the promoter. However, this result may require additional experiments to explain whether metabolic and epigenetic compensatory mechanisms are involved.
Sterol regulatory element-binding transcription factor 1 (Srebf1) is a transcription factor involved in adipocyte differentiation and the synthesis of cholesterol and fatty acids that plays a central role in energy homeostasis by promoting glycolysis, lipogenesis, and adipogenesis [13]. However, up to now, no studies have previously examined the epigenetic regulation of its promoter. Although no changes were induced in Srebf1 promoter methylation as a result of the obesogenic diet, the shift to a chow diet tended to reduce the methylation of CpG site 23. More studies are needed to ascertain whether the magnitude of this change could have metabolic implications. One of the genes which expression is directly regulated by Srebf1 is Fasn. Fasn in an enzyme that catalyzes the synthesis of long-chain fatty acids from acetyl-CoA and malonyl-CoA and is one of the rate-limiting steps in de novo lipogenesis. Changes in Fasn methylation levels have been previously reported in Wistar rats as a result of the intake of an obesogenic diet in epididymal adipose tissue [31]. Our findings also show a hypomethylating effect of HFS diet in CpG site 1, whereas the shift to a chow diet increased the methylation levels of CpGs 5 and 15–16. Multiple cycles of fasting–refeeding result in a remarkable increase in Srebf1 protein in parallel to that in fatty acid synthase mRNA and protein levels [27]. It is possible that the pivotal role that Srebf1 and Fasn plays in the increase of the lipogenic potential of adipose tissue after repeated dieting may be mediated by epigenetic alterations in the promoters of the genes. The data obtained in this study consolidate Fasn as a gene whose promoter is highly sensitive to epigenetic mechanisms of gene expression regulation due to dietary and metabolic factors.
Aqp7 is a protein that facilitates glycerol and water transport abundantly expressed in adipocytes [33]. Its mRNA levels decrease by feeding and increase by fasting, in parallel with plasma glycerol levels [33]. Although a CpG island has been located in its promoter, no studies have analyzed its epigenetic regulation. In our study, we did not find any relevant changes in the promoter methylation of this gene due to the diet.
Regulation of gene expression by DNA methylation is crucial for defining cellular identities and coordinating organism-wide developmental programs in many organisms. Although it has generally been thought that methylation is a stable epigenetic mark and that changes in methylation are unidirectional through time (cells becoming increasingly methylated as they move through the differentiation process), new data suggest that active DNA demethylation occurs in cancer, neuronal, and embryonic stem cell lines [54]. This raises the possibility that methylation might in fact be bidirectional and that there might be an as yet undiscovered, active mechanism that performs demethylation. However, DNA methylation changes are locus specific and relatively modest in magnitude [5]. All the methylation changes observed in metabolic studies are of small magnitude, but some of them have been correlated with changes in gene expression, at least in a subset of genes [20, 51].
In this study, we have shown that a shift to a normocaloric diet may reverse the methylation levels of some CpG sites located in several promoters that were previously affected by the intake of a HFS diet. There are several hypotheses that must be considered. As the shift to a control diet reverses the HFS-induced hypertrophy of adipocytes and they are again similar to those of control rats, it is easy to think that their DNA methylation patterns are again similar, that is, the adipocyte size distribution may be involved in the differential DNA methylation observed. Also, the shift to a chow diet changes the amount of energy, fat, and fiber, as well as the type of fats of the diet. Some of these factors could be responsible of modifying the DNA methylation levels of some of the analyzed genes. Finally, several metabolic syndrome traits that were dysregulated by the intake of the HFS diet are normalized after the shift to the chow diet (i.e., liver steatosis, serum lipid profile). Although not analyzed in this work, something similar should occur with different hormones such as leptin or insulin and with the elevation of chemokine and cytokine expression usually associated to adipose tissue enlargement [48]. Some of these metabolic changes could influence the DNA methylation profile of the genes analyzed.
The main limitation of the current work is the lack of RNA from the same adipocytes, whose DNA has been analyzed. This analysis could clarify whether the alterations observed in DNA methylation are actually associated with changes in gene expression.
References
Alibegovic AC, Sonne MP, Hojbjerre L, Bork-Jensen J, Jacobsen S, Nilsson E, Faerch K, Hiscock N, Mortensen B, Friedrichsen M, Stallknecht B, Dela F, Vaag A (2010) Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am J Physiol Endocrinol Metab 299:E752–763
Allison DB, Pi-Sunyer FX (1995) Obesity treatment: examining the premises. Endocr Pract 1:353–364
Amengual-Cladera E, Llado I, Gianotti M, Proenza AM (2012) Sex differences in the effect of high-fat diet feeding on rat white adipose tissue mitochondrial function and insulin sensitivity. Metabolism 61:1108–1117
Bird A (2007) Perceptions of epigenetics. Nature 447:396–398
Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A (2012) DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 47:633–647
Bondia-Pons I, Boque N, Paternain L, Santamaria E, Fernandez J, Campion J, Milagro F, Corrales F, Martinez JA (2011) Liver proteome changes induced by a short-term high-fat sucrose diet in wistar rats. J Nutrigenet Nutrigenomics 4:344–353
Boque N, Campion J, Paternain L, Garcia-Diaz DF, Galarraga M, Portillo MP, Milagro FI, Ortiz de Solorzano C, Martinez JA (2009) Influence of dietary macronutrient composition on adiposity and cellularity of different fat depots in Wistar rats. J Physiol Biochem 65:387–395
Bouchard L, Rabasa-Lhoret R, Faraj M, Lavoie ME, Mill J, Perusse L, Vohl MC (2010) Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am J Clin Nutr 91:309–320
Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, Perron P, Brisson D (2010) Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 33:2436–2441
Brons C, Jacobsen S, Nilsson E, Ronn T, Jensen CB, Storgaard H, Poulsen P, Groop L, Ling C, Astrup A, Vaag A (2010) Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J Clin Endocrinol Metab 95:3048–3056
Brug J (2007) The European charter for counteracting obesity: a late but important step towards action. Observations on the WHO-Europe ministerial conference, Istanbul, November 15–17, 2006. Int J Behav Nutr Phys Act 4:11
Campion J, Milagro FI, Martinez JA (2009) Individuality and epigenetics in obesity. Obes Rev 10:383–392
Chen J, Yang XJ, Xia D, Wegner J, Jiang Z, Zhao RQ (2008) Sterol regulatory element binding transcription factor 1 expression and genetic polymorphism significantly affect intramuscular fat deposition in the longissimus muscle of Erhualian and Sutai pigs. J Anim Sci 86:57–63
Clifton P (2012) Effects of a high protein diet on body weight and comorbidities associated with obesity. Br J Nutr 108(Suppl 2):S122–129
Cordero P, Campion J, Milagro FI, Goyenechea E, Steemburgo T, Javierre BM, Martinez JA (2011) Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J Physiol Biochem 67:463–470
Cordero P, Gomez-Uriz AM, Campion J, Milagro FI, Martinez JA (2012) Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr 8:105–113
Fontaine KR (2001) Weight loss and health-related quality of life. Am J Manag Care 7:926–927
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Galarraga M, Campión J, Muñoz-Barrutia A, Boqué N, Moreno H, Martínez JA, Milagro F, Ortiz-de-Solórzano C. (2012) Adiposoft: an automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res 53:2791–2796
Gervin K, Vigeland MD, Mattingsdal M, Hammero M, Nygard H, Olsen AO, Brandt I, Harris JR, Undlien DE, Lyle R (2012) DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PLoS Genet 8:e1002454
Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK, Alisch RS, Morley R, Visscher PM, Craig JM, Saffery R (2012) Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res 22:1395–1406
Hambly C, Speakman JR (2005) Contribution of different mechanisms to compensation for energy restriction in the mouse. Obes Res 13:1548–1557
Jacobsen SC, Brons C, Bork-Jensen J, Ribel-Madsen R, Yang B, Lara E, Hall E, Calvanese V, Nilsson E, Jorgensen SW, Mandrup S, Ling C, Fernandez AF, Fraga MF, Poulsen P, Vaag A (2012) Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 55:3341–3349
Jiang M, Zhang Y, Liu M, Lan MS, Fei J, Fan W, Gao X, Lu D (2011) Hypermethylation of hepatic glucokinase and L-type pyruvate kinase promoters in high-fat diet-induced obese rats. Endocrinology 152:1284–1289
Jousse C, Parry L, Lambert-Langlais S, Maurin AC, Averous J, Bruhat A, Carraro V, Tost J, Letteron P, Chen P, Jockers R, Launay JM, Mallet J, Fafournoux P (2011) Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: implication for the understanding of metabolic syndrome. FASEB J 25:3271–3278
Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM (2002) Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 346:393–403
Kochan Z (2003) Increased lipogenic potential of rat adipose tissue after repeated dieting—the role of SREBP-1 transcription factor. Cell Mol Biol Lett 8:901–909
Lemonnier D (1972) Effect of age, sex, and sites on the cellularity of the adipose tissue in mice and rats rendered obese by a high-fat diet. J Clin Invest 51:2907–2915
Ling C, Del Guerra S, Lupi R, Ronn T, Granhall C, Luthman H, Masiello P, Marchetti P, Groop L, Del Prato S (2008) Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51:615–622
Logue J, Thompson L, Romanes F, Wilson DC, Thompson J, Sattar N (2010) Management of obesity: summary of SIGN guideline. BMJ 340:c154
Lomba A, Martinez JA, Garcia-Diaz DF, Paternain L, Marti A, Campion J, Milagro FI (2010) Weight gain induced by an isocaloric pair-fed high fat diet: a nutriepigenetic study on FASN and NDUFB6 gene promoters. Mol Genet Metab 101:273–278
Lomba A, Milagro FI, Garcia-Diaz DF, Marti A, Campion J, Martinez JA (2010) Obesity induced by a pair-fed high fat sucrose diet: methylation and expression pattern of genes related to energy homeostasis. Lipids Health Dis 9:60
Maeda N (2012) Implications of aquaglyceroporins 7 and 9 in glycerol metabolism and metabolic syndrome. Mol Aspects Med 33:665–675
Martinez JA (2000) Body-weight regulation: causes of obesity. Proc Nutr Soc 59:337–345
Martinez JA, Cordero P, Campion J, Milagro FI (2012) Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc Nutr Soc 71:276–283
Milagro FI, Campion J, Cordero P, Goyenechea E, Gomez-Uriz AM, Abete I, Zulet MA, Martinez JA (2011) A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J 25:1378–1389
Milagro FI, Campion J, Garcia-Diaz DF, Goyenechea E, Paternain L, Martinez JA (2009) High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem 65:1–9
Milagro FI, Mansego ML, De Miguel C, Martínez JA (2012) Dietary factors, epigenetic modifications and obesity outcomes: Progresses and perspectives. Mol Aspects Med. doi:10.1016/j.mam.2012.06.010
Mirhashemi F, Scherneck S, Kluth O, Kaiser D, Vogel H, Kluge R, Schurmann A, Neschen S, Joost HG (2011) Diet dependence of diabetes in the New Zealand Obese (NZO) mouse: total fat, but not fat quality or sucrose accelerates and aggravates diabetes. Exp Clin Endocrinol Diabetes 119:167–171
Misra A, Khurana L (2008) Obesity and the metabolic syndrome in developing countries. J Clin Endocrinol Metab 93:S9–30
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14 Spec No 1:R47–58
Nixon JP, Zhang M, Wang C, Kuskowski MA, Novak CM, Levine JA, Billington CJ, Kotz CM (2010) Evaluation of a quantitative magnetic resonance imaging system for whole body composition analysis in rodents. Obesity (Silver Spring) 18:1652–1659
Orzano AJ, Scott JG (2004) Diagnosis and treatment of obesity in adults: an applied evidence-based review. J Am Board Fam Pract 17:359–369
Park LK, Friso S, Choi SW (2012) Nutritional influences on epigenetics and age-related disease. Proc Nutr Soc 71:75–83
Paternain L, Batlle MA, De la Garza AL, Milagro FI, Martínez JA, Campión J (2012) Transcriptomic and epigenetic changes in the hypothalamus are involved in an increased susceptibility to a high-fat–sucrose diet in prenatally stressed female rats. Neuroendocrinology. doi:10.1159/000341684
Paternain L, de la Garza AL, Batlle MA, Milagro FI, Martínez JA, Campión J (2012) Prenatal stress increases the obesogenic effects of a high-fat–sucrose diet in adult rats in a sex-specific manner. Stress. doi:10.3109/10253890.2012.707708
Pinnick KE, Karpe F (2011) DNA methylation of genes in adipose tissue. Proc Nutr Soc 70:57–63
Sato M, Kawakami T, Kondoh M, Takiguchi M, Kadota Y, Himeno S, Suzuki S (2010) Development of high-fat-diet-induced obesity in female metallothionein-null mice. FASEB J 24:2375–2384
Sookoian S, Rosselli MS, Gemma C, Burgueno AL, Fernandez Gianotti T, Castano GO, Pirola CJ (2010) Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 52:1992–2000
Sun L, Yang Z, Jin F, Zhu XQ, Qu YC, Shi XH, Wang L (2006) The Gly482Ser variant of the PPARGC1 gene is associated with type 2 diabetes mellitus in northern Chinese, especially men. Diabet Med 23:1085–1092
Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, Calonne E, Volkmar U, Igoillo-Esteve M, Naamane N, Del Guerra S, Masini M, Bugliani M, Marchetti P, Cnop M, Eizirik DL, Fuks F (2012) DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 31:1405–1426
Weyer C, Walford RL, Harper IT, Milner M, MacCallum T, Tataranni PA, Ravussin E (2000) Energy metabolism after 2 y of energy restriction: the biosphere 2 experiment. Am J Clin Nutr 72:946–953
Widiker S, Karst S, Wagener A, Brockmann GA (2010) High-fat diet leads to a decreased methylation of the Mc4r gene in the obese BFMI and the lean B6 mouse lines. J Appl Genet 51:193–197
Zhang Y, Chen FQ, Sun YH, Zhou SY, Li TY, Chen R (2011) Effects of DNMT1 silencing on malignant phenotype and methylated gene expression in cervical cancer cells. J Exp Clin Cancer Res 30:98
Acknowledgments
The authors thank the “Línea Especial” (LE/97, University of Navarra, Spain) for financial support. The technical assistance of Almudena Aguado and Alexandra Claire Simpson is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure 1
a Nucleotide sequence of the CpG island in the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a) promoter region showing individual CpG dinucleotides. b The effect of HFS diet intake and shifting to a chow diet in the methylation levels of individual CpG dinucleotides in the Ppargc1a promoter. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and DMS post hoc test. Different letters (a, b, c) indicate significant differences between groups of at least p < 0.05. Ppargc1a, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet; Chr., chromosome; NA, not available (PDF 114 kb)
Figure 2
a Nucleotide sequence of the CpG island in the leptin promoter region showing individual CpG dinucleotides. b The effect of HFS diet intake and shifting to a chow diet in the methylation levels of individual CpG dinucleotides in the leptin promoter. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and DMS post hoc test. Different letters (a, b) indicate significant differences between groups of at least p < 0.05. t indicates a trend to significance. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet; Chr., chromosome; NA, not available (PDF 147 kb)
Figure 3
a Nucleotide sequence of the CpG island in the sterol regulatory element-binding transcription factor 1 (Srebf1) promoter region showing individual CpG dinucleotides. b The effect of HFS diet intake and shifting to a chow diet in the methylation levels of individual CpG dinucleotides in the Srebf1 promoter. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and DMS post hoc test. Different letters (a, b) indicate significant differences between groups of at least p < 0.05. t indicates a trend to significance. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet; Chr., chromosome; NA, not available (PDF 131 kb)
Figure 4
a Nucleotide sequence of the CpG island in the Fatty acid synthase (Fasn) promoter region showing individual CpG dinucleotides. b The effect of HFS diet intake and shifting to a chow diet in the methylation levels of individual CpG dinucleotides in the Fasn promoter. Data are mean ± SEM. Statistical analyses were performed using One-way ANOVA test and DMS post hoc test. Different letters (a, b) indicate significant differences between groups of at least p < 0.05. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet; Chr., chromosome; NA, not available (PDF 135 kb)
Figure 5
a Nucleotide sequence of the CpG island in the Aquaporin 7 (Aqp 7) promoter region showing individual CpG dinucleotides. b The effect of HFS diet intake and shifting to a chow diet in the methylation levels of individual CpG dinucleotides in the Aqp 7 promoter. Data are mean ± SEM. Statistical analyses were performed using one-way ANOVA test and no statistically significant changes were observed. HFS, high-fat–sucrose; rest, animals shifted from HFS to chow diet; Chr., chromosome; NA, not available (PDF 111 kb)
Rights and permissions
About this article
Cite this article
Uriarte, G., Paternain, L., Milagro, F.I. et al. Shifting to a control diet after a high-fat, high-sucrose diet intake induces epigenetic changes in retroperitoneal adipocytes of Wistar rats. J Physiol Biochem 69, 601–611 (2013). https://doi.org/10.1007/s13105-012-0231-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-012-0231-6