
Abstract
Sirtuins (Sirt) are a family of NAD+ dependent histone deacetylase (HDAC) proteins implicated in aging, cell cycle regulation, and metabolism. These proteins are involved in the epigenetic modification of neuromodulatory proteins after strokevia acetylation/deacetylation. The specific role of Sirt3, a mitochondrial sirtuin, in post-stroke injury has been relatively unexplored. Using male Sirt3 knockout (KO) mice and wild-type littermates (WT), we show that Sirt3 KO mice show significant neuroprotection at 3 days after ischemia/reperfusion (I/R) or stroke injury. The deacetylation activity of Sirt3, measured as the amount of reduced acetylated lysine, was increased after stroke. Stroke-induced increases in liver kinase 1 (LKB1) activity were also reduced in KO mice at 3 days after stroke. On further investigation, we found that the levels of Sirt1, another important member of the Sirtuin family, were increased in the brains of Sirt3 KO mice after stroke. To determine the translational relevance of these findings, we then tested the effects of pharmacological inhibition of Sirt3. We found no benefit of Sirt3 inhibition despite clear evidence of deacetylation. Overall, these data suggest that Sirt3 KO mice show neuroprotection by a compensatory rise in Sirt1 rather than the loss of Sirt3 after stroke. Further analysis reveals that the beneficial effects of Sirt1 might be mediated by a decrease in LKB1 activity after stroke. Finally, our data clearly demonstrate the importance of using both pharmacological and genetic methods in pre-clinical stroke studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is a leading cause of death and disability in the USA [1]. Ischemia-induced impairment of energy metabolism is a critical factor contributing to brain injury and recovery [2, 3]. Besides the well-established mechanisms of cell death (i.e., excitotoxicity, calcium overload, oxidative stress, acute inflammation, and apoptosis), cellular disruption of the acetylation of histones and other proteins has recently been recognized as a common feature in many neurodegenerative disorders [4]. Given the profound effect of histone acetylation on gene expression, recent research has evaluated the use of histone deacetylase (HDAC) inhibitors in stroke and traumatic brain injury [5]. These agents exert neuroprotective effects by raising histone acetylation levels, suppressing transcription factors (e.g., p53), reducing inflammatory gene expression (e.g., IL-1b, COX), increasing extracellular glutamate clearance, or by stabilizing mitochondrial integrity [6, 7]. While these studies have focused mainly on class I and II families of HDAC inhibitors, little is known regarding HDAC III (the sirtuins) inhibition in the context of ischemia.
Sirtuins (Sirt) are NAD+ dependent HDAC proteins implicated in a variety of cellular functions including aging, cell cycle regulation, and cellular metabolism [8]. Among the seven known human Sirtuins, Sirt3 has emerged as a key regulator of several important proteins involved in mitochondrial energy metabolism. For example, loss of Sirt3 increases glycolytic metabolism, allowing metabolically challenged cells to survive longer [9, 10]. Thus, the loss of Sirt3 might increase glycolytic metabolism and help maintain the survival of vulnerable neurons. In addition to Sirt3, Sirt1 is another prominent sirtuin found mainly in the nucleus. Sirt1 and Sirt3 coordinate to increase cellular energy stores and ultimately maintain cellular energy homeostasis [11]. Several reports suggest that both Sirt1 and Sirt3 facilitate the transcriptional repression of nuclear genes encoding proteins involved in sensing metabolic stress or the deacetylation of non-histone proteins such as liver kinase B1 (LKB1) also known as serine/threonine kinase 11 [12, 13]. LKB1 is a nuclear enzyme that is activated by deacetylation in addition to conventional activation by the 14-3-3 family proteins [14]. Deacetylation of LKB1 promotes its translocation from the nucleus to the cytosol and thus facilitates its activation [15, 16] following I/R injury [17]. Therefore, we hypothesized that the loss of Sirt3 would be beneficial in stroke, potentially mediated through inhibition of LKB1 activation.
Materials and Methods
Animals and Ischemic Stroke
Sirt3 KO (129-Sirt3tm1.1Fwa/J) and WT (129S1/SvImJ) mice were purchased from the Jackson Laboratories to initiate an in-house breeding colony. A total of 164 global Sirt3 KO or littermate male WT mice were randomly divided and subjected to stroke, out of which a total of 153 were used in the final analysis. The details of the number of mice used in each individual experiment are given in the figure legends of the respective experiment. Focal transient cerebral ischemia was induced in young male mice (2–3 months, 20–25 g) for 60 min with a right middle cerebral artery occlusion (MCAo) as described previously [18]. Rectal temperatures were maintained at approximately 37 °C during surgery and ischemia with an automated temperature control feedback system. Sham-operated animals underwent the same surgical procedure, except that the suture was not advanced into the internal carotid artery. Successful occlusion was confirmed by laser Doppler reflecting < 15% residual MCA flow, with the return of flow to 85% of baseline upon reperfusion. All analysis was performed blinded to surgical condition and genotype.
India Ink Vascular Staining
WT and KO mice were anesthetized (isoflurane) and perfused with PBS followed by 4% paraformaldehyde and India ink (50% India ink, 5% FeSO4in PBS). The brains were carefully harvested and visualized using a digital camera after brief refrigeration (Panasonic HC V770).
Drug Treatment
The Sirt3 inhibitor AGK7 (sc-204281) selectively inhibits Sirt3, Sirt1, and Sirt2 (IC50 = > 5, > 50, and > 50 μM for Sirt3, Sirt1, and Sirt2, respectively). An initial dose–response curve was performed with AGK7 (0.15, 0.5, 1.5 mg/kg, i.p.). A single dose of the drug was administered 3 h after MCAo (n = 6 per treatment group). Mice were sacrificed after 6 h and/or 3 days after stroke for either histological or biochemical analysis.
Infarct Analysis
For histological assessment, all mice were euthanized at 3 days after stroke with avertin overdose (i.p). Transcardial perfusion was performed with cold PBS followed by 4% paraformaldehyde; the brain was post-fixed for 24 h and placed in cryoprotectant (30% sucrose). The brains were cut into 30-μm-thick free-floating sections on a freezing microtome, and every eighth slice was stained with cresyl violet to evaluate ischemic cell damage. The images were digitalized, and infarct measurements were analyzed by a blinded investigator using the software (Sigma Scan Pro5) as previously described [18]. Infarct volumes were presented as a percentage volume (percentage of contralateral structures with correction for edema).
Behavioral Testing
Prior to surgery, a baseline behavioral reference was generated for each behavioral test except neurological scoring. Animals were tested on each behavioral task again 3 days after stroke. Testing was performed at a fixed time in the morning (09:00–11:00) by investigators blinded to the experimental design. All equipment and surfaces were cleaned with 70% ethanol before and after testing each animal. Mice were tested in the following order: neurological scoring, open field, and rotarod.
Gross Behavioral Assessment and Neurological Deficit Score
Body weight was monitored 1 day before and every day following ischemia. Rectal temperatures were recorded during ischemia (immediately after removal from a feedback controlled surgical heating pad) and immediately before sacrifice. Neurological deficits were assessed at 3 days after stroke using the scoring system from 0 (no deficit) to 4 (severe deficit) as in [19] with minor modifications [18].
Open Field
The open field test measures spontaneous locomotor activity in a novel environment. Mice were individually placed in an open field chamber (15″ × 15″) in a dark room. Locomotor activity was quantified as the total number of beam breaks by mice using a computer-operated PAS-Open Field system (San Diego Instruments, San Diego, CA). Anxiety levels were measured by taking the percentage of the total beam breaks in the center of the chamber versus the total (peripheral + central). Each testing session lasted 10 min, and the data was collected at 300-second intervals. The sum of intervals was used to analyze the data.
Rotarod
Mice were placed on a rotating cylindrical rod accelerating from 2 rotations per minute (rpm) to 20 rpm, over a span of 5 min. Subjects were given two trials each on the rotarod on the day − 2, − 1, + 1,+ 2, and + 3 respective to stroke. A 20-min break was given between trials. The latency to fall from the rotating rod was recorded for each trial (in seconds), and the mean latency was used for comparison between groups.
Measurement of Total Glutathione
Glutathione was measured in whole tissue lysate obtained from the perilesional frontal cortex at 3 days after stroke using a protocol supplied by the vendor (Cayman, Ann Arbor, MI) to determine reduced glutathione (GSH) content in the lysate (nmol/mg protein).
Hemorrhagic Transformation
Brain slices were taken at the same distance from bregma (0.5 mm anterior to bregma) and three sections/animal (n = 4) were analyzed in the area of the infarct. Hemorrhagic transformation was calculated based on the area of oxidized hemoglobin present in the ischemic hemisphere. Using Image J software (National Institutes of Health, MD, USA), each image was adjusted to grayscale and the threshold adjusted such that only the areas of oxidized hemoglobin showed signal. This area was divided by the area of the ipsilateral hemisphere and multiplied by 100 to obtain the percent of the hemisphere containing oxidized hemoglobin that was a result of hemorrhagic transformation.
Protein Expression and Western Blot Analysis
Brain samples were obtained at 6 and 72 h (or 3 days) after stroke or sham surgery and were immediately dissected into the right ipsilateral (R; ischemic) and left (L; non-ischemic) contralateral hemispheres. The perilesional frontal cortical region of ipsilateral hemisphere was separated and homogenized with Dounce homogenizers in cold HEPES lysis buffer (250 mM sucrose, 10 mM HEPES (pH 7.5), 1.5 mM MgCl2, 15 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1% Triton X PMSF, phosphatase, and protease inhibitor cocktail) as previously described for whole cell lysate [20]. A total 30 μg of protein quantified by BCA kit (Thermo Scientific, Rockford, IL) was loaded into each well and resolved on 4–15% SDS gels and transferred to a PVDF (polyvinylidene difluoride) membrane. Sirt3, Sirt1, pLKB1, LKB1, acetylated lysine (Ac-Lys), (all from Cell Signaling Technology Danvers, MA), and Actin (Sigma-Aldrich Corp, St. Louis, MO) were detected with their respective primary antibodies. All blots were blocked with either 5% non-fat dry milk or 4% BSA for 1 h at room temperature and incubated overnight with primary antibodies at 4 °C. Secondary antibodies were either goat anti-rabbit IgG 1:10,000 (GE Healthcare Bio-Sciences Pittsburgh, PA) or goat anti-mouse IgG 1:10,000 (GE Healthcare Bio-Sciences Pittsburgh, PA). SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL) was used for signal detection. Densitometry (n = 4–6 per group) was performed with Image J software.
Statistics
All the data were analyzed and expressed as mean ± S. E. M. Behavioral and other tests comprising three or more groups were analyzed by one-way ANOVA with a Bonferroni post hoc test to correct for multiple comparisons or Student’s t test, except for neurologic deficit scores, which were analyzed by Mann–Whitney U test. In a two-factor interaction “group versus time points,” a two-way ANOVA with repeated measures was used followed by a Bonferroni post hoc correction. A probability value of p < 0.05 was considered to be statistically significant. Investigators performing and analyzing behavioral tests and other analyses were blinded to treatment and surgical conditions.
Results
Cerebral Vascular Anatomy Is similar In Both Sirt3 KO and WT mice
To rule out the possibility of inherent differences in perfusion territory in Sirt3 KO versus WT mice, we performed large vessel staining in naïve mice using India ink. There were no apparent differences in the structure of the major cerebral arteries or posterior communicating artery anastomosis (n = 5, Fig. 1a). Trans-temporal laser Doppler analysis of cortical blood flow dynamics showed no difference between Sirt3 KO and littermate WT mice during and after MCAo (Fig. 1b).

a Macroscopic analysis of the cerebral arterial vasculature shows no differences between Sirt KO and WT. Representative image of India ink staining in WT and KO mice shows branching of the internal carotid artery, anastomosis of internal carotid and basilar territories (n = 5/group). b Cortical perfusion during ischemia assessed by laser Doppler flowmetry (%LDF reduction) shows the reduction in perfusion during ischemia, and recovery of blood flow during reperfusion was not different between genotypes
Genetic Deletion of Sirt3 Is Neuroprotective
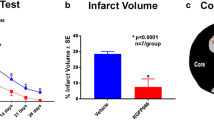
Sirt3 KO mice had significantly reduced hemispheric infarct size (p < 0.05) compared to littermate WT control (37.72 ± 5.21 vs. 52.8 ± 6.76; n = 8–9/group) (Fig. 2a). This was reflected in the observed neurological deficit score (NDS) where a significant reduction (p < 0.05) was found at 3 days after stroke in Sirt3 KO mice (Fig. 2b). Sirt3 KO and WT mice showed no differences in general locomotor activity after stroke as evidenced by equivalent total beam breaks in the open field (Fig. 3a). However, KO mice spent more time (p < 0.05) in the central zone of the open field, indicative of a reduction in the post-stroke anxiety phenotype (Fig. 3b). Sirt3 KO mice spent significantly more time (p < 0.05) on an accelerating rotarod (Fig. 3c), indicating they had less deficits in balance and motor coordination. Sirt3 KO mice also had increased (p < 0.05) amount of reduced glutathione (GSH) after stroke (Fig. 3d) suggesting that KO mice had a superior antioxidant defense system compared to WT after stroke.

Sirt3 KO mice have smaller infarcts at 3 days after stroke. a Representative images of CV staining are shown. b KO mice show a significant reduction (*p < 0.05 vs. WT, Student’s t test) in infarct volume in both the cortex and total ipsilateral hemisphere. c Congruent with infarct volume, the neurological deficit score (ND score) was also significantly reduced (*p < 0.05 vs. WT, Mann–Whitney U test) in KO mice (n, KO = 9; WT = 8)

Sirt3 KO mice show improved behavioral recovery and elevated glutathione (reduced) levels 3 days after stroke. a Both KO and WT mice show no change in exploratory activity as measured by total beam breaks. b KO mice show significantly less (*p < 0.05 vs. WT, Student’s t test) anxiety-like behavior after stroke. c KO mice show increased (*p < 0.05 vs. WT, Student’s t test) latency to fall (i.e., spent more time on a rotarod suggesting improved balance and coordination in KO mice after stroke. d Interestingly, reduced glutathione (GSH) level was higher (*p < 0.05 vs. WT, Student’s t test) in KO mice, suggesting enhanced antioxidant defense in these mice (n, KO = 6; WT = 7)
Sirt3 Activity, but Not Expression, Changed with Stroke
Sirt3 protein expression was the same in sham and stroke mice (Fig. 4a) at different time points after stroke. However, Ac-Lys levels were significantly decreased (p < 0.05) after stroke (Fig. 4a). Further, a progressive loss of lysine acetylation was noted with increasing reperfusion duration in both WT and KO mice after stroke. Basal levels of acetylated lysine were higher in KO mice (Fig. 4c).

Sirt3 regulates protein acetylation after stroke. a Western blot images for Sirt3 and Ac-lysine expression using whole cell lysate from Sirt3 KO and WT mice after stroke. b Temporal analysis of Sirt3 protein expression after sham, 60 min/6 h or 60 min/72 h (3 days) after stroke showed no change in total Sirt3 protein levels. c However, temporal analysis of Ac-lys indicates two important findings; first, the level of Ac-lysine protein was higher (*p < 0.05 vs. WT, Student’s t test) in KO compared to their respective time point controls, and second, the amount of Ac-lysine decreases progressively after stroke (*p < 0.05 a one-way ANOVA). A two-way ANOVA found a significant interaction between stroke surgery and genotype [F (2,10) = 31.39 p < 0.001] as well as the main effect of both stroke [F (2,10) = 206 p < 0.001] and genotype [F (1,5) = 246.9 p < 0.001] on Ac-lys expression (n = 4mice/group/time point; graphs show mean + S.E.M.)
Sirt1 Expression Is Upregulated in Sirt3 KO Mice After Stroke
Given that two of the seven sirtuins (Sirt1 and Sirt3) are structural homologs, overlapping and redundant functions may compensate for isotype deletion [11]. Both Sirt1 and Sirt3 are ubiquitously expressed in the brain especially in neurons [21,22,23,24]. We found that Sirt1 and Sirt3 are co-expressed in the same cells in the brain (Supplementary Fig. 1). Sirt3 is widely expressed in NeuN-positive neurons (Supplementary Fig. 2) and Iba-1-positive microglia (Supplementary Fig. 4) but is minimally expressed in GFAP-positive astrocytes (Supplementary Fig. 3). We also examined the expression of Sirt1 in Sirt3 KO mice after stroke (Fig. 5a–c). Sirt1 expression was significantly increased (p < 0.05) in Sirt3 KO mice after stroke (Fig. 5b). This suggests that elevated Sirt1 expression might partially compensate for the loss of Sirt3 under stress conditions.

Effect of stroke on Sirt3 and Sirt1 and their downstream target LKB1. a Representative western blot image of Sirt3, Sirt1, and LKB1. b Sirt3 KO mice show complete absence of Sirt3 protein. Stroke does not increase its total expression in WT mice. c Sirt1 expression was low in sham Sirt3 KO mice but increased at 3 days after stroke in these mice (*p < 0.01 vs. WT, Student’s t test). Two-way ANOVA showed a significant interaction between stroke and genotype [F 1, 12 = 99.24 p < 0.001] on Sirt1 expression. d pLKB1 levels were significantly reduced (*p < 0.01 vs. WT, Student’s t test) in Sirt3 KO mice at 3 days after stroke (n = 4mice/group; graphs show mean + S.E.M.)
Sirt3-Mediated Deacetylation Activates the Serine/Threonine Kinase LKB1
Nearly all cells that express Sirt3 also express LKB1 in the brain (Supplementary Fig. 5). Deacetylation of LKB1 increases its activation by phosphorylation at Ser428 [16]. Phosphorylated LKB1 (pLKB1) then further activates numerous downstream targets [25]. We found that stroke-induced increased expression of pLKB1 was reduced (p < 0.05) in KO mice (Fig. 5d). These findings suggest that Sirt3 might be involved in the deacetylation-mediated phosphorylation of LKB1.
Effect of Pharmacological Inhibition of Sirt3 in WT Mice
Next, we tested the effect of the Sirt3 inhibitor in both Sirt3 KO and WT mice. We first optimized the dose of inhibitor based on its effects on lysine acetylation (Fig. 6). We saw a ceiling effect at a 0.5-mg/kg dose of the inhibitor above which no additional increase in lysine acetylation was seen. We also saw a significant reduction in Sirt3 expression after inhibitor treatment, but the effect was not dose-dependent (Fig. 6). Sirt3 inhibition reduced Sirt1 expression in WT mice after stroke (Fig. 6).Unexpectedly, the Sirt3 inhibitor did not decrease infarct volume at a dose of 0.15 or 0.5 mg/kg (Fig. 7a, b). Moreover, a higher dose (1.5 mg/kg) of the inhibitor unexpectedly increased infarct volume (51.8 ± 6.87 vs. 62.56 ± 7.89) and significantly increased hemorrhagic transformation. We observed a similar trend of increase in hemorrhagic transformation after treatment (1.5 mg/kg) in Sirt3 KO mice (Fig. 7c). The Sirt3 inhibitor also reduced expression of pLKB1 at a dose of 0.15 or 0.5 mg/kg but unexpectedly restored its expression at a dose of 1.5 mg/kg (Fig. 8) to the level seen in the vehicle-treated group. These data suggest that pharmacological inhibition of Sirt3 may be ineffective or even detrimental due to off-target effects on Sirt1. This suggests that the neuroprotective effects seen in Sirt3 KO mice may be mediated by a compensatory increase in Sirt1 levels rather than the loss of Sirt3.

Effect of pharmacological inhibition of Sirt3 on Sirt1 expression, Sirt3 activity, and expression. Western blot and accompanying histogram show that doses of 0.5 and 1.5 mg/kg i.p. of Sirt3 inhibitor “AGK7” significantly decrease (*p < 0.05 vs veh; one-way ANOVA) Sirt3 activity [measured in terms of a decrease in acetylated lysine (Ac-lys) expression] and expression of both Sirt1 and Sirt3 at 3 days after stroke in WT mice (*p < 0.05 vs. veh, one-way ANOVA; n = 6/group/time point, graphs show mean + S.E.M.)

Effect of Sirt3 inhibitor AGK7 on infarct volume and hemorrhagic transformation. a Representative images of cresyl violet (CV) staining are shown. b No dose-dependent effect of Sirt3 inhibition was seen in WT mice at 3 days after stroke. However, a higher dose (1.5 mg/kg i.p.) paradoxically increased infarct volume by ~ 10–15% as compared to vehicle and other doses in WT mice after stroke. c Both the lower and intermediate doses (0.15 and 0.5 mg/kg i.p., respectively) of inhibitor did not cause hemorrhagic transformation in WT mice; however, the higher dose (1.5 mg/kg. i.p.) led to significant (*p < 0.05 vs. veh; one-way ANOVA) hemorrhagic transformation in both WT and KO mice suggesting its off-target effect at this dose (n = 6–8/mice group; graphs show mean + S.E.M.)

Effect of the Sirt3 inhibitor “AGK7” on LKB1 phosphorylation (i.e., activation). Representative image and accompanying histogram show that the lower and intermediate dose (0.15 and 0.5 mg/kg i.p., respectively) of inhibitor showed a significant reduction in LKB1 phosphorylation (*p < 0.05 vs. veh; one-way ANOVA). However, a higher dose (1.5 mg/kg i.p.) paradoxically increased LKB1 phosphorylation at 3 days after stroke in WT mice (n = 6/time point/group; graphs show mean + S.E.M.)
Discussion
The present study provides evidence for neuroprotection in male Sirt3 KO mice after ischemic brain injury. We also found decreased activity of LKB1 in Sirt3 KO mice after stroke, which coincides with reduced ischemic damage. Using both genetic deletion and pharmacological inhibition of Sirt3, we demonstrate that the deacetylation activity of Sirt3, measured by reduced levels of acetylated lysine [26], increased after stroke. In our first set of experiments, we showed that genetic deletion of Sirt3 led to acute neuroprotection and reduced behavioral deficits after stroke. Interestingly, we observed no change in total Sirt3 protein levels after ischemia/reperfusion (I/R) injury. However, Sirt3 activity (as measured by deacetylation activity) increases after stroke. At baseline (sham animals), lysine acetylation was higher in KO mice, which suggests reduced deacetylation activity in the absence of Sirt3. This comparison also highlights the significant contribution of Sirt3 in brain deacetylation, consistent with previous observations [27].
Although little is known about how deacetylation of lysine activates/deactivates other cellular proteins, previous reports suggest Sirt3-mediated deacetylation may re-activate metabolic enzymes that help cope with the need for increased ATP production through β-oxidation in mitochondria during ischemic stress [28]. One recent study has shown that I/R injury triggers Sirt3-dependent deacetylation of ceramide synthases and this elevation of ceramide leads to increased ROS generation and an exacerbation of brain injury [29]. Several other targets of Sirt3-mediated deacetylation have been reported [30] and may play important roles in stroke outcome. We found an increase in the active form of LKB1 in the brain (i.e., pLKB1) after stroke as compared to sham in WT mice, but an unexpected reduction of pLKB1 in Sirt3 KO mice after stroke. This observation suggests that Sirt3 regulates LKB1 activity, which is consistent with prior findings in non-neural tissues [31]. Therefore, the neuroprotection seen in Sirt3 KO mice may be partially mediated by this reduction in LKB1 activation. A reduction in pLKB1 and its downstream targets (e.g., AMPK) leads to neuroprotection in ischemic stroke, and this may be the mechanism by which injury is reduced in Sirt3 KO mice [32, 33].
This study also found that Sirt1 is upregulated in Sirt3 KO mice after stroke, which we examined after our pharmacological attempts to lower Sirt3 which led to an exacerbation of ischemic injury. Both Sirt1 and Sirt3 share high levels of sequence homology and substrate specificity and have many common functions and targets, but differ in their relative subcellular location [11]. Therefore, we thought a compensatory increase in Sirt1, which has been shown to be protective in ischemic injury [34], might also influence stroke outcome in the Sirt3 KO mice. Although little is known about the overlapping roles of Sirt3 and Sirt1, genetic compensation is a common mechanism seen in many “loss of function” models [35,36,37,38] and can be a major issue in constitutive knockout models that is often ignored. To confirm that inhibition of Sirt3 is a potential therapeutic target, we utilized a relatively specific pharmacological inhibitor of Sirt3. Inhibition of Sirt3 in WT mice not only decreased Sirt3 but also reduced Sirt1 expression. Given that both Sirt3 and Sirt1 have both common and unique targets, examining total lysine acetylation does not provide specific information about their target effects. However, the reduction of total lysine acetylation seen in Sirt3 KO mice after stroke, even in the setting of increased Sirt1 expression, suggests Sirt3’s prime role is as a deacetylating enzyme.
The observance of increased post-stroke Sirt1 levels in Sirt3 KO mice prompted us to develop additional experiments to determine if it was the reduction in Sirt3 or the increase in Sirt1 that led to the neuroprotective effect in Sirt3 KO mice. Pharmacological inhibition did not reduce ischemic damage, and higher doses (1.5 mg/kg) that we confirmed restored endogenous lysine acetylation did not reduce LKB1 activity (i.e., stroke-induced elevation in pLKB1), but rather increased infarct volume and led to hemorrhagic transformation. This suggests that the drug “AGK7” had off-target effects at higher doses and led to inhibition of other “protective” Sirtuins, likely Sirt1. Acetylation/deacetylation of LKB1 occurs on multiple lysine residues sites [15]; it is possible that Sirt1 and Sirt3 act on different but specific lysine residues on downstream targets. This needs to be explored further in future studies. Unlike our observations in Sirt3 KO mice where we saw a compensatory increase in Sirt1, we did not see any increase in Sirt1 expression after pharmacological inhibition of Sirt3 with AGK7, rather we saw a dose-dependent reduction in Sirt1 expression. This is consistent with the hypothesis that the neuroprotection seen in Sirt3 KO mice is due to the compensatory increase in Sirt1 rather than the loss of Sirt3. Importantly, this study highlights the need of using both genetic and pharmacological methods in translational stroke research.
Conclusions/Limitations
Sirt3 KO mice show neuroprotection from ischemic stroke. However, acute pharmacological inhibition of Sirt3 did not recapitulate our genetic findings. This prompted us to examine potential off-target effects in both models. Sirt3 KO mice had a compensatory increase in Sirt1, and this increase in Sirt1 likely led to neuroprotection in Sirt3 KO mice after stroke. This is consistent with our findings after using a Sirt3 inhibitor, which at high doses, worsened ischemic damage, in parallel with an increase in off-target inhibition of Sirt1. Despite showing several interesting observations, there are some limitations to this study as we did not explore in detail the effects of the compensatory increase of Sirt1 in Sirt3 KO mice on targets like LKB1 activation. We did not examine the effect of Sirt3 deletion or inhibition at chronic time points after stroke, as these would likely be similar to the 3-day outcomes. Although our study shows that targeting Sirt3 might not be a promising neuroprotective strategy, nevertheless, the enhanced expression of Sirt1 confirms that this may be a good therapeutic target for further development.
References
Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart Disease and Stroke Statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360. https://doi.org/10.1161/CIR.0000000000000350.
Robbins NM, Swanson RA. Opposing effects of glucose on stroke and reperfusion injury: acidosis, oxidative stress, and energy metabolism. Stroke. 2014;45(6):1881–6. https://doi.org/10.1161/STROKEAHA.114.004889.
Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14(5):497–500. https://doi.org/10.1038/nm1735.
Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab. 2013;33(9):1335–46. https://doi.org/10.1038/jcbfm.2013.93.
Gibson CL, Murphy SP. Benefits of histone deacetylase inhibitors for acute brain injury: a systematic review of animal studies. J Neurochem. 2010;115(4):806–13. https://doi.org/10.1111/j.1471-4159.2010.06993.x.
Baltan S, Morrison RS, Murphy SP. Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics. 2013;10(4):798–807. https://doi.org/10.1007/s13311-013-0201-x.
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321(3):892–901. https://doi.org/10.1124/jpet.107.120188.
Bassett SA, Barnett MP. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients. 2014;6(10):4273–301. https://doi.org/10.3390/nu6104273.
Lu X, Bennet B, Mu E, Rabinowitz J, Kang Y. Metabolomic changes accompanying transformation and acquisition of metastatic potential in a syngeneic mouse mammary tumor model. J Biol Chem. 2010;285(13):9317–21. https://doi.org/10.1074/jbc.C110.104448.
Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19(3):416–28. https://doi.org/10.1016/j.ccr.2011.02.014.
Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92(3):1479–514. https://doi.org/10.1152/physrev.00022.2011.
Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21(8):920–8. https://doi.org/10.1101/gad.1527307.
Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103(27):10224–9. https://doi.org/10.1073/pnas.0603968103.
Bai Y, Zhou T, Fu H, Sun H, Huang B. 14-3-3 interacts with LKB1 via recognizing phosphorylated threonine 336 residue and suppresses LKB1 kinase function. FEBS Lett. 2012;586(8):1111–9. https://doi.org/10.1016/j.febslet.2012.03.018.
Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–35. https://doi.org/10.1074/jbc.M805711200.
Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285(5):3133–44. https://doi.org/10.1074/jbc.M109.077271.
Khan M, Dhammu TS, Matsuda F, Singh AK, Singh I. Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy. BMC Neurosci. 2015;16(1):42. https://doi.org/10.1186/s12868-015-0179-x.
Verma R, Friedler BD, Harris NM, McCullough LD. Pair housing reverses post-stroke depressive behavior in mice. Behav Brain Res. 2014;269:155–63. https://doi.org/10.1016/j.bbr.2014.04.044.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. https://doi.org/10.1161/01.STR.20.1.84.
Verma R, Harris NM, Friedler BD, Crapser J, Patel AR, Venna V, et al. Reversal of the detrimental effects of post-stroke social isolation by pair-housing is mediated by activation of BDNF-MAPK/ERK in aged mice. Sci Rep. 2016;6(1):25176. https://doi.org/10.1038/srep25176.
Huang W, Huang Y, Huang RQ, Huang CG, Wang WH, Gu JM, et al. SIRT3 expression decreases with reactive oxygen species generation in rat cortical neurons during early brain injury induced by experimental subarachnoid hemorrhage. Biomed Res Int. 2016;2016:8263926–9. https://doi.org/10.1155/2016/8263926.
Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404(1):1–13. https://doi.org/10.1042/BJ20070140.
Sakamoto J, Miura T, Shimamoto K, Horio Y. Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett. 2004;556(1–3):281–6. https://doi.org/10.1016/S0014-5793(03)01444-3.
Weir HJ, Murray TK, Kehoe PG, Love S, Verdin EM, O'Neill MJ, et al. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One. 2012;7(11):e48225. https://doi.org/10.1371/journal.pone.0048225.
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–8. https://doi.org/10.1016/j.cub.2003.10.031.
Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433(3):505–14. https://doi.org/10.1042/BJ20100791.
Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27(24):8807–14. https://doi.org/10.1128/MCB.01636-07.
Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864(10):1372–401. https://doi.org/10.1016/j.bbapap.2016.06.007.
Novgorodov SA, Riley CL, Keffler JA, Yu J, Kindy MS, Macklin WB, et al. SIRT3 deacetylates ceramide synthases: implications for mitochondrial dysfunction and brain injury. J Biol Chem. 2016;291(4):1957–73. https://doi.org/10.1074/jbc.M115.668228.
Kupis W, Palyga J, Tomal E, Niewiadomska E. The role of sirtuins in cellular homeostasis. J Physiol Biochem. 2016;72(3):371–80. https://doi.org/10.1007/s13105-016-0492-6.
Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP. Mitochondrial SIRT3 and heart disease. Cardiovasc Res. 2010;88(2):250–6. https://doi.org/10.1093/cvr/cvq250.
McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280(21):20493–502. https://doi.org/10.1074/jbc.M409985200.
Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38(11):2992–9. https://doi.org/10.1161/STROKEAHA.107.490904.
Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69(2):360–74. https://doi.org/10.1002/ana.22236.
Jackson JG, Pereira-Smith OM. Primary and compensatory roles for RB family members at cell cycle gene promoters that are deacetylated and downregulated in doxorubicin-induced senescence of breast cancer cells. Mol Cell Biol. 2006;26(7):2501–10. https://doi.org/10.1128/MCB.26.7.2501-2510.2006.
Bhattacharjee P, Paul S, Banerjee M, Patra D, Banerjee P, Ghoshal N, et al. Functional compensation of glutathione S-transferase M1 (GSTM1) null by another GST superfamily member, GSTM2. Sci Rep. 2013;3(1):2704. https://doi.org/10.1038/srep02704.
Kreiner G. Compensatory mechanisms in genetic models of neurodegeneration: are the mice better than humans? Front Cell Neurosci. 2015;9:56. https://doi.org/10.3389/fncel.2015.00056.
Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, et al. Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc Natl Acad Sci U S A. 2015;112(10):3122–7. https://doi.org/10.1073/pnas.1417498112.
Funding
This work was supported by National Institutes of Health grants R01NSO77769 (to Louise D McCullough) and an AHA postdoctoral fellowship 14POST20380612 (to Rajkumar Verma).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All animal protocols were approved by the University’s Institutional Animal Care and Use Committee at UConn Health, Farmington, CT, and were performed in accordance with National Institutes of Health guidelines.
Electronic supplementary material
ESM 1
(DOCX 13128 kb)
Rights and permissions
About this article

Cite this article
Verma, R., Ritzel, R.M., Crapser, J. et al. Evaluation of the Neuroprotective Effect of Sirt3 in Experimental Stroke. Transl. Stroke Res. 10, 57–66 (2019). https://doi.org/10.1007/s12975-017-0603-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-017-0603-x