
Abstract
Polarized microglia play a dual (beneficial/detrimental) role in neurological diseases. However, the status and the factors that modulate microglia polarization in intracerebral hemorrhage (ICH) remain unclear. In the present study, we investigated the role of protease-activated receptor-1 (PAR-1, a thrombin receptor) in ICH-induced microglia polarization in mice. Male wild-type (WT) and PAR-1 knockout (PAR-1 KO) mice received an infusion of 30-μL autologous blood or saline into the right basal ganglia. Mice were euthanized at different time points and the brains were used for Western blotting and immunohistochemistry. Some mice had magnetic resonance imaging. We found that ICH induced microglia activation and polarization. M1 phenotypic markers were markedly increased and reached a peak as early as 4 h, remained high at 3 days and decreased 7 days after ICH. M2 phenotypic markers were upregulated later than M1 markers reaching a peak at day 1 and declining by day 7 after ICH. PAR-1 was upregulated after ICH and expressed in the neurons and microglia. ICH induced less brain swelling and neuronal death in PAR-1 KO mice, and this was associated with less M1 polarization and reduced proinflammatory cytokine levels in the brain. In conclusion, these results suggest that polarized microglia occur dynamically after ICH and that PAR-1 plays a role in the microglia activation and polarization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Increasing evidence from preclinical and clinical studies suggests that inflammation contributes to secondary brain injury after intracerebral hemorrhage (ICH) [1, 2]. Such inflammation is characterized by the accumulation and activation of inflammatory cells and release of inflammatory mediators within the hemorrhagic brain. Inflammatory cells include blood-derived leukocytes and macrophages, resident microglia, astrocytes, and mast cells [1, 3].
Microglia act as guardians of neuronal survival and function under homeostatic condition as well as various pathologic conditions in the brain [3, 4]. Despite their distinct origin, microglia share many phenotypic and functional properties with blood-derived macrophages [5, 6]. Phagocytic microglia obtain an amoeboid morphology, becoming indistinguishable from infiltrating activated macrophages after activation [1, 7]. Accumulating evidence suggests that the inflammatory response mediated by microglia/macrophage has an important role in ischemic and hemorrhagic brain injury [1, 2, 7–9].
Evidence suggests that microglia/macrophages are highly plastic cells that can assume diverse phenotypes and engage different functional programs in response to specific micro-environmental signals [10, 11]. In particular, lipopolysaccharide and proinflammatory cytokine interferon-γ promote “classically activated” microglia/macrophage (M1 phenotype) that release proinflammatory mediators, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and reactive oxygen species, causing damage to healthy cells and tissues [12]. In contrast, cytokines such as interleukin-4 and interleukin-13 can induce an “alternative activated” microglia/macrophage (M2 phenotype) that generate anti-inflammatory cytokines (e.g., transforming growth factor beta (TGF-β), interleukin-10 (IL-10)) and contribute to neuro-protective properties [9, 13–15].
Microglia/macrophage activation and M1/M2 polarization have been well characterized in several types of acute central nervous system injury, including traumatic brain injury [16, 17], spinal cord injury [10], and ischemic stroke [11, 18]. Previous studies have revealed that the activation of microglia/macrophages occurs in the hemorrhagic brain and results in inflammatory injury after ICH [1, 7, 19–21]. However, the detailed mechanisms underlying microglia activation and polarization after ICH remain largely unclear.
Previous studies have demonstrated that protease-activated receptor-1 (PAR-1), a member of G protein-coupled protease-activated receptors, is involved in inflammatory responses, mostly with pro-inflammatory roles. Over-activation of PAR-1 is detrimental to neuronal survival and related to brain injury in various neurodegenerative diseases including ICH [22–26]. However, whether PAR-1 is involved in microglia/macrophage activation and polarization, and the precise mechanisms whereby PAR-1 promotes inflammation after ICH are unknown.
In this study, we used a well-established mouse model to investigate the status of microglia/macrophage activation and polarization after ICH. We also examined the role of PAR-1 in ICH-induced inflammation and brain injury.
Materials and Methods
Animal Preparation and Intracerebral Injection
All animal procedures were approved by the University Committee on Use and Care of Animals, University of Michigan. A total of 62 adult male C57BL/6 wild-type (WT) mice (Jackson Laboratory, Bar Harbor, ME, USA) and 48 male PAR-1 knockout (PAR-1 KO) mice (University of Michigan Breeding Core) at age of 2–3 months were used in this study. Mice were housed at a controlled temperature under a 12-h light-dark cycle, and all animals were allowed free access to food and water.
Intracerebral injections were performed as previously described [27]. Briefly, mice were anesthetized with ketamine (90 mg/kg, intraperitoneally, Abbott Laboratories, Chicago, IL, USA) and xylazine (5 mg/kg intraperitoneally, Lloyd Laboratories, Shenandoah, IA, USA). Rectal temperature was maintained at 37.5 °C by a feedback-controlled heating pad. The right femoral artery was catheterized to monitor arterial blood pressure and blood glucose levels, and to sample blood for intracerebral autologous blood infusion. The mice were then positioned in a stereotaxic frame (Model 500, Kopf Instruments, Tujunga, CA, USA), and a cranial burr hole (1 mm) was drilled near the right coronal suture 2.5 mm lateral to the midline. A 26-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates 0.2 mm anterior, 3.5 mm ventral, and 2.5 mm lateral to the bregma). Either 30-μL autologous whole blood, TFLLR-NH2 (a PAR-1 agonist, Sigma, St. Louse, MO, USA) or the same volume of saline was infused at 2 μL/min by a microinfusion pump (Harvard Apparatus Inc., South Natick, MA, USA). After injection, the needle remained in position for 10 min to prevent reflux and then it was gently removed. The burr hole was filled with bone wax, and the skin incision was closed with suture.
Experimental Groups
There were two sets of experiments in this study. In the first set, WT and PAR-1 KO mice had 30 μL of autologous blood or saline infusion (control) and were euthanized at 4 h, and days 1, 3, and 7 (n = 6 per group per time point) after intracerebral infusion. Some mice had serial magnetic resonance imaging (MRI) at days 1 and 3 after intracerebral infusion (n = 9 per group at day 1, n = 6 per group at day 3). The harvested brains were used for immunohistochemistry (n = 3 per group, per time point) and Western blotting assays (n = 3 per group, per time point). In the second set, WT mice had either 30 μL of TFLLR-NH2 (30 nmol, n = 7) or saline (n = 7). The brains were harvested at 24 h.
Magnetic Resonance Imaging
Mice were anesthetized with 2 % isoflurane/air mixture throughout the MRI examination. MRI was performed at days 1 and 3 in a 7.0-T varian MR scanner (183-mm horizontal bore; Varian, Palo Alto, CA, USA) at the Center for Molecular Imaging of University of Michigan. The MRI included a T2 fast spin-echo sequence (repetition time/echo time = 4000/60 ms) using a field of view of 20 × 20 mm, matrix of 256 × 256 mm, and 25 coronal slices (0.5 mm thickness). Images were preserved as 256 × 256 pixel pictures for analysis of brain swelling with NIH ImageJ (Wayne Rasband, National Institutes of Health, USA). All measurements were repeated three times and the mean value used.
Western Blot Analysis
Mice were anesthetized before undergoing intracardiac perfusion with 0.1 mol/L phosphate-buffered saline. The brains were then removed and a 2-mm thick coronal brain slice was cut approximately 2 mm from the frontal pole. The slice was separated into the ipsilateral and contralateral basal ganglia. Western blot analysis was performed as previously described [28, 29]. Briefly, 50-μg protein samples were separated by sodium dodecyl sulfate polyacrymide gel electrophoresis and transferred onto a hybond-C pure nitrocellulose membrane (Amersham, Pittsburgh, PA, USA). The membranes were blocked in Carnation nonfat milk and probed with the primary and secondary antibodies. The primary antibodies were rabbit anti-PAR1 IgG (Abcam, Cambridge, MA, USA, 1:1000 dilution), rabbit anti-cluster of differentiation 16 (CD16) IgG (Abcam, 1:1000 dilution), mouse anti-inducible nitric oxide synthase (iNOS) IgG (Abcam, 1:1000 dilution), goat anti-chitinase 3 like protein 3 (YM-1) IgG (Abcam, 1:1000 dilution), rabbit anti-cluster of differentiation 206 (CD206) IgG (Abcam, 1:1000 dilution), goat anti-matrix metallopeptidase 9 (MMP9) IgG (R&D, Minneapolis, MN, USA, 1:1000 dilution), rabbit anti-TNF-α IgG (Abcam, 1:1000 dilution), rabbit anti-IL-1β IgG (Abcam, 1:1000 dilution), rabbit anti-TGF-β IgG (Abcam, 1:1000 dilution), rabbit anti-IL-10 IgG (Abcam, 1:1000 dilution), and mouse anti-glyceraldehyde 3 phosphate dehydrogenase (GAPDH) IgG (Cell signaling, 1:500,000 dilution). The secondary antibodies were goat anti-rabbit IgG, rabbit anti-goat IgG, and goat anti-mouse IgG (Bio-Rad, Hercules, CA, USA, 1:2000 dilution). The antigen-antibody complexes were visualized with the ECL chemiluminescence system (Amersham) and exposed to Kodak X-OMAT film (Rochester, NY, USA). The relative densities of bands were analyzed with the NIH ImageJ.
Immunohistochemistry and Immunofluorescence Double Labeling
Immunohistochemistry and immunofluorescence double staining was performed as described previously [28]. Briefly, mice were reanesthetized and subjected to intracardiac perfusion with 4 % paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). Brains were then harvested and kept in 4 % paraformaldehyde for 24 h, before immersion in 30 % sucrose for 3 to 4 days at 4 °C. After embedding in a mixture of 30 % sucrose and OCT (Sacura Finetek, Inc., Torrance, CA, USA), 18-μm sections were taken on a cryostat and then preserved at −80 °C.
The avidin-biotin complex technique was used in the immunohistochemistry. The primary antibodies were rabbit anti-PAR1 IgG (Abcam, Cambridge, MA, USA, 1:200 dilution), rabbit anti-CD16 IgG (Abcam, 1:200 dilution), mouse anti-iNOS IgG (Abcam, 1:200 dilution), goat anti-YM-1 IgG (Abcam, 1:100 dilution), and rabbit anti-CD206 IgG (Abcam, 1:200 dilution). For immunofluorescence double labeling, the primary antibodies were polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) IgG (Millipore, Billerica, MA, USA, 1:400 dilution), polyclonal mouse anti-neuronal-specific nuclear protein (NeuN) IgG (Millipore, Billerica, MA, USA, 1:200 dilution), and those for PAR-1, CD16, CD206 and iNOS. The secondary antibodies were Alexa Fluro 488-conjugated donkey anti-rabbit mAb (Invitrogen, Grand Island, NY, USA, 1:500 dilution), Alexa Fluro 594-conjugated donkey anti-mouse mAb (Invitrogen, 1:500 dilution), and Alexa Fluro 594-conjugated donkey anti-goat mAb (Invitrogen, 1:500 dilution). The double labeling was analyzed using a fluorescence microscope (Olympus, BX51). Omitting primary antibody was used as negative control.
Fluoro-Jade C Staining
For assessment of neuronal degeneration, mounted brain sections were stained with Fluoro-Jade C [30, 31]. Briefly, slides were dried on a slide warmer at 50 °C for 30 min. Then, the following steps were performed in the dark, sections were treated with 0.06 % potassium permanganate for 15 min. After a brief rinse in double-distilled water, the sections were stained with 0.001 % Fluoro-Jade C in 1 % acetic acid for 25 min on a shaker. Sections were rinsed 3 × 1 min in double-distilled water and followed by air-drying on the slide warmer at 50 °C for 8 min. Slides were then cleared in xylene and mounted.
DNA Polymerase I-Mediated Biotin-dATP Nick-Translation (PANT) Labeling
PANT labeling was performed to detect DNA strand breaks. Sections were permeabilized with 1 % Triton X-100 for 30 min and quenched with 2 % hydrogen peroxide for 20 min followed by three phosphate-buffered saline (PBS) washes. They were then incubated in a moist-air chamber at 37 °C for 60 min in the PANT solution. The reaction was terminated by washing the slides twice in PBS. After washing for 5 min in PBS containing bovine serum albumin (0.5 mg/mL), slides were incubated with streptavidin-horseradish peroxidase in PBS containing bovine serum albumin for 90 min at room temperature. Detection of the biotin-streptavidin-peroxidase complex was carried out by incubation with diaminobenzidine solution.
Cell Counting
Three high-power images (×40 magnification) were taken of the ipsilateral basal ganglia around the hematoma using a digital camera. Fluoro-Jade C and PANT-positive cells were counted in three perihematomal areas from each brain section by a blinded observer and expressed as cells per square millimeter.
Statistical Analysis
All data in this study are presented as means ± S.D. Data were analyzed by Student’s t test for single comparisons or ANOVA for multiple comparisons. Statistical significance was set at p < 0.05.
Results
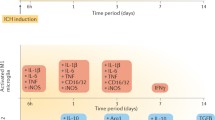
Time Course of M1/M2 Microglia/Macrophage Polarization After ICH
Immunohistochemistry showed an increase of CD16, iNOS (markers of M1 phenotypic microglia/macrophage, Fig. 1a, b), CD206, and YM-1 (markers of M2 phenotypic microglia/macrophage, Fig. 1c, d) positive cells in the ipsilateral basal ganglia after ICH compared to sham controls in WT mice. Western blot analysis showed that the levels of CD16 and iNOS (Fig. 1a, b) were markedly increased and reached at peak as early as 4 h in ipsilateral basal ganglia after ICH, remained at a high level until 3 days, and decreased at 7 days after ICH. While the M2 phenotypic microglia/macrophage was activated slower than M1 phenotypic microglia/macrophage, time course showed that the levels of CD206 and YM-1 were upregulated at 4 h in the ipsilateral basal ganglia after ICH, reached at peak on the first day, and decreased on 7 days after ICH (Fig. 1c, d).

Immunoreactivity and time course of protein levels of cluster of differentiation 16 (CD16, a), inducible nitric oxide synthase (iNOS, b), cluster of differentiation 206 (CD206, c), and chitinase 3 like protein 3 (YM-1, d) in the ipsilateral basal ganglia after injection of 30 μL autologous blood or saline into the right basal ganglia. Values are mean ± SD, n = 3 for each group. #p < 0.05 vs. day 1 and day 3; *p < 0.01 vs. other groups. Scale bar = 20 μm
PAR-1 Expression After ICH
Brain PAR-1 levels were increased after ICH (Suppl. Fig. I). Western blot analysis showed that the protein levels of PAR-1 were markedly elevated as early as 4 h in ipsilateral basal ganglia, reached a peak on the 1 day, and decreased on 7 days after ICH (Fig. 2a). Double labeling showed that PAR-1 immunoreactivity co-localized with NeuN (a neuronal marker), CD16 (an M1 phenotypic microglia/macrophage marker), and CD206 (an M2 phenotypic microglia/macrophage marker) immunoreactivity. It did not co-localize with GFAP (an astrocyte marker) (Fig. 2b).

Time course of protein levels of PAR-1 (a) in the ipsilateral basal ganglia after injection of 30 μL autologous blood or saline into the right basal ganglia. Values are mean ± SD, n = 3 for each group, #p < 0.05 vs. day 1 or day 3 groups, or *p < 0.01 vs. other group; and immunofluorescent double labeling (b) of PAR1 with neuron marker (NeuN), astrocyte marker (GFAP), M1 phenotype (CD16), and M2 phenotype (CD206). Scale bar = 20 μm
ICH-Induced Less Brain Swelling and Neuronal Death in PAR-1 KO mice
Hematoma volume, measured by T2* lesion volume, showed no difference between PAR1 KO and WT mice at 24 h after ICH (15.2 ± 2.1 vs. 14.9 ± 3.0 mm3, p > 0.05). The % brain swelling, ([ipsilateral − contralateral hemisphere] × 100 / contralateral hemisphere) as assessed by T2 MRI, was significantly less in PAR-1 KO mice compared to WT mice (day 1 8.6 ± 1.1 vs. 13.0 ± 0.4 %, p < 0.05; day 3 6.7 ± 0.5 vs. 9.2 ± 0.6 %, p < 0.05; Fig. 3a). The levels of MMP9 were markedly increased in the ipsilateral basal ganglia after ICH and were significantly lower in PAR-1 KO compared with WT mice (MMP9/GAPDH 0.45 ± 0.12 vs. 0.94 ± 0.16 in WT, p < 0.05; Fig. 3b). In addition, in PAR-1 KO mice, there were fewer Fluoro-Jade C-positive cells (237 ± 124 vs. 895 ± 310 cell/mm2 in WT mice, p < 0.01; Fig. 3c) and PANT-positive cells (35 ± 14 vs. 109 ± 26 cell/mm2 in WT mice, p < 0.01; Fig. 3d) in the ipsilateral basal ganglia 1 day after ICH.

a T2-weighted magnetic resonance imaging (MRI) showing brain swelling [(ipsilateral-contralateral hemisphere) / contralateral hemisphere × 100 %] at days 1 and 3 after intracerebral hemorrhage (ICH). Values are mean ± SD, n = 6, #p < 0.05 vs. wild-type (WT) group. b Protein levels of MMP-9 at 1 day after ICH. Values are mean ± SD, n = 3 for each group, *p < 0.01 vs. control group, #p < 0.05 vs. wild-type group after ICH. c Fluoro-Jade C-positive cells in ipsilateral basal ganglia of wild-type and PAR-1 KO mice at 1 day after ICH. Values are mean ± SD, n = 3, *p < 0.01 vs. wild-type group, scale bar = 20 μm. d PANT-positive cells in ipsilateral basal ganglia of wild-type and PAR-1 KO mice at 1 day after ICH. Values are mean ± SD, n = 3, *p < 0.01 vs. wild-type group, scale bar = 20 μm
ICH Induced Less M1, but Not M2 Polarization in PAR-1 KO Mice
Immunohistochemistry showed a marked increase in the numbers of CD16 and iNOS-labeled microglia/macrophages in the ipsilateral basal ganglia 1 day after ICH in WT mice compared to sham controls. That increase was lower in PAR-1 KO mice (Suppl. Fig. II). Western blot analysis confirmed those findings. CD16 and iNOS protein levels were markedly upregulated in the ipsilateral basal ganglia of WT mice compared to saline controls 1 day after ICH (Fig. 4a, b), but that upregulation was significantly lower in PAR-1 KO mice (CD16/GAPDH 1.01 ± 0.19 vs. 1.72 ± 0.21 in WT mice, p < 0.05; Fig. 4a; iNOS/GAPDH 0.50 ± 0.12 vs. 1.10 ± 0.21 in WT mice, p < 0.05; Fig. 4b).

Protein levels of cluster of differentiation 16 (CD16, a) and inducible nitric oxide synthase (iNOS, b) in the ipsilateral basal ganglia 1 day after injection of 30-μL autologous blood or saline into the right caudate. Values are mean ± SD, n = 3 for each group, *p < 0.01 vs. control groups and #p < 0.05 vs. wild-type group
Immunohistochemistry and Western blots of CD206 (a marker of microglia/macrophage M2 phenotype) both showed an upregulation in the ipsilateral basal ganglia 1 day following ICH compared to saline controls (Suppl. Fig. III). However, there was no significant difference between PAR-1 KO and WT mice (p > 0.05; Fig. 5a). Similar results were found for another M2 marker, YM-1. Again, there was an upregulation in YM-1 in the ipsilateral basal ganglia after ICH in WT animals compared to sham controls as assessed by immunohistochemistry and Western blot, but there was no significant difference between PAR-1 KO and WT mice (p > 0.05; Fig. 5b).

Protein levels of cluster of differentiation (CD206, a) and chitinase 3 like protein 3 (YM-1, b) in the ipsilateral basal ganglia 1 day after injection of 30-μL autologous blood or saline into the right caudate. Values are mean ± SD, n = 3 for each group, *p < 0.01 vs. control groups
Effects of PAR-1 KO on Pro- and Anti-Inflammatory Cytokines Expression After ICH
ICH upregulated TNF-α and IL-1β protein levels in the ipsilateral basal ganglia at the first day, and the protein level of TNF-α and IL-1β was significantly lower in PAR-1 KO compared with WT mice (TNF-α/GAPDH 0.52 ± 0.11 vs. 0.88 ± 0.11 in WT, p < 0.05, Fig. 6a; IL-1β/GAPDH 1.49 ± 0.21 vs. 2.01 ± 0.06 in WT, p < 0.05; Fig. 6b). However, ICH-induced upregulation of TGF-β and IL-10 was not significantly different between PAR-1 KO and WT mice (p > 0.05; Fig. 6c, d).

Protein levels of tumor necrosis factor-α (TNF-α, a), interleukin-1β (IL-1β, b), interleukin-10 (IL-10, c), and transforming growth factor-β (TGF-β, d) in the ipsilateral basal ganglia 1 day after injection of 30-μL autologous blood or saline into the right caudate. Values are mean ± SD, n = 3 for each group, *p < 0.01 or #p < 0.05 vs. control groups, #p < 0.05 vs. wild-type group
PAR-1 Agonist Activated M1 Microglia/Macrophages
Intracaudate injection of TFLLR-NH2 activated microglia/macrophages. CD16 levels in the ipsilateral basal ganglia were upregulated 24 h after TFLLR-NH2 injection (Fig. 7a). However, TFLLR-NH2 did not affect YM-1 levels (Fig. 7b).

Protein levels of CD 16 (a) and YM-1 (b) in the ipsilateral basal ganglia 1 day after injection of 30 μL TFLLR-NH2 or saline into the right caudate. Values are mean ± SD, n = 4 for each group, #p < 0.05 vs. the saline control group
Discussion
The major findings of our present study were as follows: (1) ICH induces microglia activation and polarization. M1 phenotype microglia/macrophages are markedly increased, reach a peak as early as 4 h in the ipsilateral basal ganglia, remain high at 3 days, and decrease at 7 days after ICH. Meanwhile, the activation of M2 phenotypic microglia/macrophages is delayed, reaching a peak at day 1. (2) PAR-1 is involved in ICH-induced brain injury and modulates microglia/macrophage polarization. Thus, PAR-1-deficient mice had less M1 phenotypic microglia/macrophage activation, inflammation, DNA damage and neuron death, and brain swelling after ICH. Future studies should determine whether PAR-1 affects ICH-mediated neurological deficits.
Increasing evidence suggests that inflammatory mechanisms are involved in stroke-induced brain injury, and microglia/macrophage activation is thought to play critical pathophysiological roles [1, 7–9, 32]. Stroke-induced over-activation of microglia/macrophages can amplify inflammatory response, aggravate blood-brain barrier disruption and neuronal damage, and represent a promising target for stroke treatment. However, selective ablation of proliferating microglia exacerbates brain injury after ischemic stroke [33], and anti-inflammation therapies have, as yet, failed to improve clinical outcomes in stroke patients. Therefore, it is likely that broad suppression of microglia/macrophages may deprive the brain of a normal physiological defense mechanism and result in undesired effects. Thus, an improved understanding of the dual beneficial and detrimental roles of microglia in brain injury and recovery is crucial for future stroke management [11].
Microglia are known as the resident macrophages of the central nervous system, contributing to brain homeostasis. Once activated, microglia/macrophages serve as a double-edged sword in the battle between neurological damage and protection. On the one hand, classically activated microglia/macrophages (M1 phenotype) can release devastating pro-inflammatory mediators (e.g., TNF-α, IL-1β, interleukin 6) and oxidative metabolites, exacerbating inflammatory damage. On the other hand, alternative activated microglia/macrophages (M2 phenotype) can secrete numerous protective and neurotrophic factors (e.g., TGF-β, IL-10, brain-derived neurotrophic factor, interleukin 1 receptor antagonist), promote brain recovery by scavenging cell debris, resolve local inflammation, and are involved in tissue remodeling [9, 34, 35]. Therefore, it is important to realize the status and function of polarized microglia activation for assessing inflammation progression and optimizing treatment. Recent research has studied microglia/macrophage activation and polarization in several neurological conditions, including brain ischemia [10, 11, 16–18]. However, the polarized microglia/macrophage activation after ICH has not been well studied [36, 37]. The present study reveals the time course of polarized microglia/macrophage activation after ICH, which should provide a solid basis for further investigation to microglia/macrophage-mediated inflammatory injury and repair after ICH.
PARs are a subfamily of G protein-coupled receptors with four members (PAR-1, PAR-2, PAR-3, and PAR-4) [38], of which PAR-1, PAR-3, and PAR-4 are initially recognized as receptors for coagulation proteases and are involved in thrombin signaling, whereas PAR-2 is activated by trypsin and tryptase [39–41]. In addition, numerous other proteinases have been shown to cleave and activate PAR1 including activated protein C, matrix metalloproteinases-1, and neutrophil elastase [42]. Once activated, the PARs are able to stimulate multiple intracellular signaling networks via classical G protein-mediated pathways and β-arrestin signaling. In addition, different receptor crosstalk mechanisms critically contribute to a high diversity of PAR signal transduction and receptor-trafficking processes that result in multiple physiological effects [43]. Increasing evidence has demonstrated that PAR-1 activation is involved in inflammatory responses, mostly pro-inflammatory, and is related to brain injury after hemorrhagic and ischemic stroke [22–24]. Thus, the second question addressed in this study was whether PAR-1 activation after ICH is involved in microglia/macrophage activation and polarization. The present study demonstrated that the activation of PAR-1 is indeed triggered within 4 h of ICH, with a peak at day 1 and a decline at day 7. The time course of PAR-1 activation is consistent with the timing of microglia/macrophage activation and polarization. PAR-1 expression was found in M1 and M2 phenotypic microglia/macrophages as well as neurons.
The results also showed that PAR-1 deficiency could inhibit M1 phenotypic microglia/macrophage activation and reduce the expression of pro-inflammatory cytokines after ICH. PAR-1 deficiency had less effect on M2 phenotypic microglia/macrophage activation and polarization after ICH. Accordingly, the expression of neurotrophic cytokines was similar between the PAR-1-deficient mice and wild-type mice. Also in support of these findings, intracaudate injection of a PAR-1 agonist increased tissue levels of CD-16, a marker of M1 phenotypic microglia/macrophages but had no effect on YM-1, a M2 marker. The present study reveals that PAR-1 activation, presumably by clot-derived thrombin, is involved in microglia/macrophage activation and polarization after ICH. PAR-1activation may contribute microglia/macrophage-mediated inflammatory brain injury, at least partially, through modulating microglia/macrophage polarization toward a skewed M1 phenotype in mice brain, amplifying the release of pro-inflammatory cytokines as well as MMP-9, an important regulator of blood-brain barrier disruption after injury [44]. This may exacerbate DNA damage and neuronal death, and aggravate brain edema and neurological deficits after ICH.
Therefore, a third question emerging is the intracellular molecular switches that might regulate PAR-1-mediated microglia/macrophage polarization. A recent breakthrough in research on macrophages has disclosed that several transcriptional regulators may serve crucial roles in switching on M1 or M2 genes and thereby promote polarization [9]. For instance, the nuclear hormone receptor peroxisome proliferator-activated receptor γ is an important transcriptional factor that mediates macrophages primed toward M2 polarization, interferon regulatory factor 4 serves as a key transcriptional factor modulating M2 polarization, whereas the interferon regulatory factor 5 and interferon regulatory factor 8 control macrophages toward M1 polarization [45, 46].
In summary, our results reveal the time course of microglia/macrophage activation and polarization after ICH in mice. That time course is consistent with PAR-1 activation, and PAR-1 deficiency inhibits M1 phenotypic microglia/macrophage activation, reduces the expression of MMP-9 and proinflammatory cytokines, ameliorates DNA damage and neuron death, and reduces brain injury after ICH. Therefore, targeting PAR-1 and its downstream interacting proteins may provide new opportunities to modulate microglia/macrophage-mediated inflammatory injury and recovery after ICH.
References
Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92(4):463–77. Pubmed Central PMCID: 2991407.
Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5(1):53–63.
van Rossum D, Hanisch UK. Microglia. Metab Brain Dis. 2004;19(3-4):393–411.
Eyo UB, Wu LJ. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013;2013:456857. Pubmed Central PMCID: 3775394.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5. Pubmed Central PMCID: 3719181.
Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399–418. Pubmed Central PMCID: 2773116.
Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. Pubmed Central PMCID: 3810327.
Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11(1):56–64. Pubmed Central PMCID: 4395497.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–44. Pubmed Central PMCID: 2788152.
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–70.
Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, et al. Alternative versus classical activation of macrophages. Pathobiology. 1999;67(5-6):222–6.
Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60(5):717–27.
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cerebral Blood Flow Metabolism. 2013;33(12):1864–74. Pubmed Central PMCID: 3851898.
Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J neurotrauma. 2015. doi:10.1089/neu.2015.4268.
Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8:174. Pubmed Central PMCID: 3251548.
Yabluchanskiy A, Sawle P, Homer-Vanniasinkam S, Green CJ, Motterlini R. Relationship between leukocyte kinetics and behavioral tests changes in the inflammatory process of hemorrhagic stroke recovery. Int J Neurosci. 2010;120(12):765–73.
Xue M, Del Bigio MR. Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000;283(3):230–2.
Wasserman JK, Zhu X, Schlichter LC. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res. 2007;1180:140–54.
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64.
Xue M, Hollenberg MD, Demchuk A, Yong VW. Relative importance of proteinase-activated receptor-1 versus matrix metalloproteinases in intracerebral hemorrhage-mediated neurotoxicity in mice. Stroke. 2009;40(6):2199–204. Epub 2009/04/11. eng.
Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, et al. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci U S A. 2003;100(22):13019–24. Pubmed Central PMCID: 240737.
Cheng Y, Xi G, Jin H, Keep RF, Feng J, Hua Y. Thrombin-induced cerebral hemorrhage: role of protease-activated receptor-1. Transl Stroke Res. 2014;5(4):472–5. Pubmed Central PMCID: 3962522.
Xi G, Strahle J, Hua Y, Keep RF. Progress in translational research on intracerebral hemorrhage: is there an end in sight? Prog Neurobiol. 2014;115C:45–63. Pubmed Central PMCID: 3961535.
Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100(4):672–8.
Xi G, Keep RF, Hua Y, Xiang J, Hoff JT. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 1999;30(6):1247–55.
Zheng M, Du H, Ni W, Koch LG, Britton SL, Keep RF, et al. Iron-induced necrotic brain cell death in rats with different aerobic capacity. Transl Stroke Res. 2015;6(3):215–23. Pubmed Central PMCID: 4425582.
Okauchi M, Xi G, Keep RF, Hua Y. Tissue-type transglutaminase and the effects of cystamine on intracerebral hemorrhage-induced brain edema and neurological deficits. Brain Res. 2009;1249:229–36. eng.
Schmued LC, Albertson C, Slikker Jr W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751(1):37–46.
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–31. Pubmed Central PMCID: 3884550.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–605.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. Pubmed Central PMCID: 2724991.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–85.
Ni W, Mao S, Xi G, Keep RF, Hua Y. Role of Erythrocyte CD47 in Intracerebral Hematoma Clearance. Stroke; a journal of cerebral circulation. 2016;47:505–11
Zhao H, Garton T, Keep RF, Hua Y, Xi G. Microglia/Macrophage Polarization After Experimental Intracerebral Hemorrhage. Transl Stroke Res. 2015;6(6):407–9. Pubmed Central PMCID: 4628553.
Luo W, Wang Y, Reiser G. Protease-activated receptors in the brain: receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res Rev. 2007;56(2):331–45.
McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A. 2007;104(13):5662–7. Pubmed Central PMCID: 1838494.
Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394(6694):690–4.
Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84(1):3–9.
Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130(3):248–82.
Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Communicat signal. 2013;11:86. Pubmed Central PMCID: 3842752.
Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50(4):329–39.
Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750–61.
Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol. 2005;161(1-2):113–22.
Acknowledgments
This study was supported by grants NS-073595, NS-079157, NS-084049, NS-090925, NS-091545 and NS-096917 from the National Institutes of Health (NIH), 973 Program-2014CB541600, and by grants 81371336, 81400942, 81400945 from National Natural Science Foundation of China (NSFC) and grants LY13H090005 from Natural Science Foundation of Zhejiang Province (ZJNSF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, NSFC, and ZJNSF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Shu Wan, Yingying Cheng, Hang Jin, Dewei Guo, Ya Hua, Richard F. Keep, and Guohua Xi declare that they have no conflict of interest.
Compliance with Ethics Requirements
All institutional and national guidelines for the care and use of laboratory animals were followed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Supplemental Figure I: Wild-type (WT) mice received an intracerebral injection of autologous whole blood or saline. At 24 hours, the brains were used to assess PAR-1 immunoreactivity (A) and protein levels (B) in the ipsilateral basal ganglia. Scale bar = 50 μm; values are means ± S.D.; n = 3 per group, *p < 0.01 vs. other groups. Supplemental Figure II: Immunoreactivity of cluster of differentiation 16 (CD16, A) and inducible nitric oxide synthase (iNOS, B) in the ipsilateral basal ganglia 1 day after injection of 30 μL autologous blood or saline into the right caudate. Scale bar=20μm. Supplemental Figure III: Immunoreactivity and protein levels of cluster of differentiation (CD206, A) and chitinase 3 like protein 3 (YM-1, B) in the ipsilateral basal ganglia 1 day after injection of 30 μL autologous blood or saline into the right caudate. Scale bar=20μm. (PDF 28266 kb)
Rights and permissions
About this article

Cite this article
Wan, S., Cheng, Y., Jin, H. et al. Microglia Activation and Polarization After Intracerebral Hemorrhage in Mice: the Role of Protease-Activated Receptor-1. Transl. Stroke Res. 7, 478–487 (2016). https://doi.org/10.1007/s12975-016-0472-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-016-0472-8