
Abstract
Notch signaling is critically involved in various biological events. Notch undergoes cleavage by the γ-secretase enzyme to release Notch intracellular domain that will translocate into nucleus to result in expression of target gene. γ-Secretase inhibitors have been developed as potential treatments for neurological degenerative diseases, but its effects against ischemic injury remain relatively uncertain. In the present study, we demonstrated that N-[N-(3, 5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), a γ-secretase inhibitor not only rescued the cerebral hypoperfusion or ischemia neonatal rats from death, reduced apoptosis in penumbra, but also reduced brain infarct size. Furthermore, DAPT elicited some morphologic hallmarks such as neurogenesis and angiogenesis that related to the brain repair and functional recovery after stroke: increased accumulations of newborn cells in the peri-infarct region with a higher fraction of them adopting immature neuronal and glial markers instead of microglial markers on 5 days, enhanced vascular densities in penumbra at 14 days, and evident regulations of the gene profiles associated with neurogenesis in penumbral tissues. The current results suggest that DAPT is a potential neuroprotectants against ischemic injury in immature brain, and future treatment strategies such as clinical trials using γ-secretase inhibitors would be an attractive therapy for perinatal ischemia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neonatal stroke has increasingly been recognized as a significant cause of mortality and long-term neurological deficits in newborns [1–3]. To ameliorate such outcomes, various experimental treatment modalities, such as hypothermia [4, 5], xenon and sevoflurane inhalation [6, 7], and hypoxic/ischemic preconditioning [8], have been offered. For further improvements in outcome, new robust neuroprotectants such as pharmacological interventions may be beneficial.
Notch signaling is critically involved in various biological events, including the maintenance of stem cells, cell fate specification, cell survival, and tissue morphogenesis [9–11]. Notch undergoes cleavage by the γ-secretase enzyme to release Notch intracellular domain (NICD) that will translocate into nucleus to result in expression of target gene. Inhibition of the γ-secretase enzyme blocks the Notch signaling pathway. Recently, γ-secretase inhibitors have been developed as potential treatments for several neurological degenerative diseases, such as Alzheimer disease [12]. However, its effects against ischemic injury remain relatively uncertain.
Ischemic stroke results from damage and death of neurons in the perfusion territory of the affected blood vessels. Notch signaling may play key roles in the cerebral ischemic injury mainly via the following ways: (1) apoptosis and inflammatory response: Notch-1, one of the Notch cell-surface receptors has been shown to endanger neurons either by modulating pathways that increase their vulnerability to apoptosis or by activating microglias, a marker for inflammatory reaction after stroke in the adult rat brain [13]; and (2) neurogenesis and angiogenesis: Notch signaling pathway engages in cell proliferations and differentiations in normal developing brain [9–11], and restricts angiogenic cell behavior to tip cells in developing segmental arteries [14]. Since increasing evidence shows that stroke-induced neurogenesis as well as angiogenesis contributes to neuroprotection and functional recovery after stroke [15–19], Notch signaling pathway may play key roles in brain repair after stroke by regulating neurogenesis and angiogenesis.
The present study was performed to explore the γ-secretase inhibitor; N-[N-(3, 5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) rescued the neonatal rats who sustained severe global hypoperfusion or focal cerebral ischemia from death, protected the neurons from apoptosis and saved the ischemic brain tissues, and elicited some morphologic hallmarks such as neurogenesis and angiogenesis in the penumbral region that related to the brain repair and functional recovery after ischemic injury in the immature brain.
Materials and Methods
Animal Handling and Ischemic Models
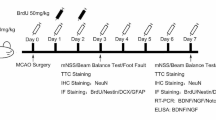
The Zhejiang University Administrative Panel of Laboratory Animal Care approved all experimental protocols. Male 10-day Sprague Dawley neonate rats (25–30 g) were anesthetized with 1.5–3 % isoflurane.
Two models were used in our experiments: (1) hypoperfusion: the bilateral common carotid arteries (CCAs) were permanently occluded; and (2) transient focal cerebral ischemia [19]: the CCA, external carotid, and pterygopalatine arteries were exposed and ligated on the left side. The left internal carotid artery (ICA) was occluded with a microsurgical clip, and an arteriotomy was made in the CCA. A 7.0-monofilament suture (Ethicon) with a rounded tip was inserted into the CCA and advanced through the ICA to the ostium to occlude the middle cerebral artery. After 1 h, the suture was removed, the wound was closed, and the rats were allowed to recover. Sham surgery was performed without artery occlusion in sham controls. Rats of hypoperfusion or transient focal cerebral ischemia were divided into three groups, respectively: (1) ischemia, (2) DAPT: DAPT (100 mg/kg) was injected (I.P.) 1 h after the occlusion, and (3) sham control.
To label newborn cells, bromodeoxyuridine (BrdU, Sigma, 50 mg/kg) was injected (I.P.) into rats 1 h after occlusion, and daily for the next 2 days.
Infarct Size Assay
For infarct size measurement, the rats were anesthetized and perfused intracardially with cold sodium phosphate-buffered saline at day 1 after occlusion. The rats were then decapitated and the brains rapidly removed and sectioned coronally at 2-mm intervals. All slices were incubated in 2 % 2,3,7-triphenyltetrazolium chloride (TTC) solution for 20 min at room temperature, fixed by 4 % paraformaldehyde solution overnight. Using a free image analysis system (Imaging J, version 1.61), the area of infarct of the two sides of each section was measured. For focal cerebral ischemia, which injuries both cortex and striatum, we normalized the infarct size to the contralateral semi-hemisphere, and an average value from all slices was presented.
Western Blotting
The peri-infarct tissues were dissected on ice [20] at day 5 after occlusion. They were then homogenized in ice-cold lysis buffer (containing 20 M Tris, pH 7.5; 150 M NaCl; 1 M ethylenediaminetetraacetic acid (EDTA); 1 M ethylene glycol tetraacetic acid (EGTA); 1 % Triton X-100; 2.5 M Na pyrophosphate; 1 M b-glycerophosphate; 1 M Na3VO4; 1.1 g/ml leupeptin; and 1 M phenylmethanesulfonyl fluoride (PMSF)). Samples were centrifuged at 14,000 rpm for 15 min. The protein concentration of the supernatant was determined using a modified Bradford assay (Sigma). Samples were normalized to equal protein concentrations. Loading buffer was added to the samples, and they were boiled for 5 min. Total protein (30 μg) was separated by electrophoresis in a 10 % polyacrylamide gel. The blots were incubated overnight at 4 °C first with the primary antibodies hairy enhancer of split (Hes) 1 (1:1000), Hes5 (1:1000), or Notch1 (1:1000) (all from Santa Cruz Biotechnology), then with peroxidase conjugated IgG (Sigma), and last with an ECL kit. Control experiments without primary antibody were negative. Optical density of the films was quantified by the free software (Image J 1.61), calculated as a value of integrated densities, and then were converted to percentage values with β-actin. All data were expressed relative to the control, representing the relative expression of the target protein.
Immunohistochemistry and Immunofluorescence Staining, Confocal Checking, and Cell Counting
These procedures were performed as previously described in detail [19, 20]. Briefly, rats were sacrificed on day 5, and the brain vibratome sections (35 μm) for BrdU-labeled cells in the ischemic cortex and ipsilateral subventricular zone (SVZ) were prepared (Fig. 1a). Every eighth section was selected for staining according to the standard protocol. Sections were immunostained with primary antibodies followed by secondary antibodies conjugated to biotin or fluorescent labels [19]. The sections allocated for BrdU staining underwent pretreatment in 2 N HCl at 37 °C for 30 min. Primary antibodies include glial fibrillary acidic protein (GFAP; 1:500; Harlan), a marker for mature astrocytes; NG2 (1:300; Chemicon), for oligodendrocytes; nestin (1:500; BD), for immature neurons and glia; NeuN (1:500; Chemicon), for mature neurons; doublecortin (DCX; 1:300; Santa Cruz Biotech), for migrating neuroblasts; BrdU (1:500; Accurate Chemicals), for dividing cells; Iba1 (1:500; Wako Pure Chemical Industries), a marker for microglias; and laminin (1:500; sigma), to label the vessels [21]. The accumulation of BrdU+ cells in the peri-infarct region and SVZ was determined using diaminobenzidine (DAB) staining, and positive cells were counted using light microscopy.

Diagrams indicate the analyzed peri-infarct, penumbra, and subventricular zone (SVZ) regions, and an outline of protocols used for microarray. a The schematic diagrams of the peri-infarct, penumbra, and SVZ regions of interest for tissue isolations or brain section preparations, and the region of interest for immunostaining qualification for blood vessels in the penumbra. The black area represents the infarct core. b Schematic outline of protocols used for microarray. c Representative array pictures from N-[N-(3, 5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT)-treated stroke rat. Each chip contained housekeeping genes (glyceraldehydes-3-phosphate dehydrogenase, GAPDH) for normalization, positive controls, negative controls, and two blank spots as additional non-specific binding controls
Cells were checked using a confocal microscope (Zeiss 510), and cell phenotypes were identified by colocalization of phenotypic markers with BrdU [19]. The relative percentage of BrdU+ cells co-labeled with each marker among the total number of BrdU+ cells and the percentage of cells expressing five markers in each group were calculated. Three views per section were subjected to the semiquantification of vessels in penumbra (Fig. 1a) by using ImageJ 1.61 with ‘Measure RGB’ Plugin. The value of each sample was normalized to the mean value of sham control group. Five sections of each brain were used in these experiments, respectively.
TUNEL
The number of cells undergoing apoptosis was assessed in penumbra by means of in situ terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining using the ApoTag Red in situ apoptosis detection kit (Millipore) according to the manufacturer’s instructions. Three views (Fig. 1a) per section were subjected to the qualification of TUNEL+ cells in penumbra by using fluorescence microscope. The total numbers of each sample were normalized to the mean value of sham control group.
RNA Preparation, Nonradioactive cDNA Microarray and Reverse Transcription, and Quantitative Real-Time Polymerase Chain Reaction
Rats were sacrificed at 5 days after focal cerebral ischemia. The tissue corresponding to the ischemic penumbra was dissected. The ischemic penumbra was defined as previously reported (Fig. 1a) [20]. Total RNA was extracted by using Trizol Reagent and purified using a spin column RNA purification kit (SuperArray Inc., Frederick, MD, USA) in accordance with the manufacturer’s procedure. To remove possible genomic DNA contamination, RNase-free DNase was used during the RNA purification steps.
The microarray experiment was performed using TrueLabeling-AMP 2.0 Kit (SuperArray) and Oligo GEArray DNA Microarray Rat Neurogenesis and Neural Stem Cell Kit (ERN-404, SuperArray) in accordance with the manufacturer’s procedure. One microgram purified RNA was used for complementary DNA (cDNA) synthesis. The biotin-16-UTP (10 mM, Roche Applied Science) was used to label the complementary RNA (cRNA), which was then purified using a spin column cRNA purification kit (SuperArray Inc.). A total of 4 μg purified biotin-labeled cRNA was used for hybridization. The array membranes were incubated with biotin-labeled probes at 60 °C for 6 h. The membranes were then washed twice at 60 °C for 5 min each. Chemiluminescent detection steps were performed by subsequent incubation with alkaline phosphatase-conjugated streptavidin and CDPStar substrate. For quantification, intensity of spots was measured by GEArray Expression Analysis Suite software (http://GEAsuite.superarray.com) and then the total intensities derived from blank spots were subtracted (see Fig. 1b, c).
By using SuperScript™ III Platinum® SYBR® Green One-Step Quantitative Real-Time PCR Kit (Invitrogen), both cDNA synthesis and polymerase chain reaction (PCR) were performed in a single tube using gene-specific primers and total RNA, whereas quantitative reverse transcription (qRT-PCR) was performed on an ABI 7500 PCR instrument. The program parameters were performed as follows: 3 min at 50 °C, 5 min at 95 °C, and then each cycle for 15 s at 95 °C, 30 s at 60 °C for 45 cycles, 1 min at 95 °C, and followed by melting curve analysis (30 s at 55 and at 95 °C). Table S1 lists primers (Invitrogen) specific for the genes examined in the present study. Each sample was tested in triplicate and the fold changes to the sham were quantified by the 2-△△CT method. Briefly, the threshold cycle (CT) values of specific transcripts were subtracted to the CT of the control gene (GAPDH), i.e., △CT; next, calculated the mean △CT of sham control group; then subtracted △CT of each sample to the mean △CT of sham control group to get the value of △△CT for each sample; and finally, used the equation (2-△△CT) to calculate the fold changes for each sample.
Statistical Analysis
All data are presented as means ± SEM. Differences between groups were assessed by ANOVA (univariate analysis of variance tests) followed by post hoc tests (LSD). Statistical significance was determined at the P < 0.05 level.
Results
DAPT Promoted Survival After Ischemia
DAPT significantly increased the percentages of survivor after ischemia. On day 5, the survival ratios were 77. 8 % (7/9) and 72.7 % (8/11) in DAPT group as well as 33.3 % (6/18) and 33.3 % (7/21) in rats without DAPT administration, under permanent hypoperfusion and transient focal ischemia conditions, respectively (Fig. 2a, b). DAPT-treated rats gained a gradual increasing of the body-weight as sham controls, but not the vehicle-treated ischemic animals that they did severely face with feeding and motor problems.

DAPT promoted survival of neonatal rats after ischemia. a Hypoperfusion was induced by the permanent occlusion of the bilateral common carotid arteries in 10-day neonatal rats. DAPT (100 mg/kg) was injected (I.P.) 1 h after the occlusion. *P < 0.05 (Chi-square). b Transient focal cerebral ischemia was induced by the occlusion of the left middle cerebral artery for 1 h. DAPT (100 mg/kg) was injected (I.P.) 1 h after the occlusion. *P < 0.05 (Chi-square)
DAPT Inhibited the Notch Signaling After Ischemia
Figure 3 shows the blots and relative density ratios of the bands in the different groups. The expression of the NICD significantly decreased in the penumbra of DAPT-treated ischemic rats in comparison with ischemic rats on day 5 after surgery. We then assessed two downstream targets within the Notch signaling pathway in the penumbra: Hes1 and Hes5. Hes1 and Hes5 were downregulated in the DAPT group on day 5 compared with the ischemia group. The blots are shown in Fig. 3b.

DAPT inhibited the Notch signaling after ischemia. a Protein expression of the Notch1 intracellular domain (NICD) and its downstream targets Hes1 and Hes5 in the three experimental groups. There is a statistically significant difference between the DAPT vs. the ischemia + vehicle group (## P < 0.001). **P < 0.001, *P < 0.05 vs. sham; ANOVA, n = 6. Values are mean ± SEM. b Representative blots of the NICD, Hes1 and Hes5 in the three groups
DAPT Reduced Infarct Size and Apoptosis After Ischemia
The results of infarct size assay indicate that a single treatment with DAPT substantially reduced infarct size under focal cerebral ischemia (Fig. 4a, b). At 1 day after ischemia, DAPT reduced 22.26 % brain infarct volume compared with the vehicle-treated ischemic rats.

DAPT reduced infarct size and apoptosis after ischemia. a, b The transient focal cerebral ischemic rats were decapitated at 24 h after occlusion and the brains rapidly removed and sectioned coronally at 2-mm intervals relative to bregma. All slices were incubated in 2 % 2,3,7-triphenyltetrazolium chloride (TTC) solution for 20 min and fixed overnight. The area of infarct of the two sides of each section was measured and then normalized the values to the contralateral semi-hemisphere, and an average value from all slices per rat was presented. a Representative infarcts stained by TTC. b DAPT (black bar) significantly decreased the infarct size compared vehicle-treated rats (white bar). Values are mean ± SEM; *P < 0.05, ANOVA, n = 6. c DAPT robustly decreased the total numbers of TUNEL+ cells in penumbra in comparison with that of ischemic rats without DAPT treatment at 24 h after surgery (## P < 0.001). **P < 0.001 vs. sham; ANOVA, n = 6. Values are mean ± SEM. d, e Representative TUNEL staining in penumbra under transient focal cerebral ischemia. Scale bars 20 μm
Furthermore, DAPT robustly decreased the total numbers of TUNEL+ cells in penumbra in comparison with that of ischemic rats without DAPT treatment on day 1 after surgery (Fig. 4c–e).
The Effects of DAPT on New Born Cell Accumulations After Stroke
For rats receiving consecutive labeling of BrdU for 3 days after stroke, the accumulated BrdU+ cell numbers were robustly increased at 5 days after stroke in the peri-infarct region after both hypoperfusion and transient focal ischemia compared with the cortex of sham rats. DAPT further increased this accumulated BrdU+ cell number in the peri-infarct region compared with the same region of ischemic rats without DAPT treatment. However, stroke resulted in a reduced-density of BrdU+ cells in the ipsilateral SVZ at day 5; such reduction was attenuated by DAPT (Fig. 5a–d).

The effects of DAPT on newborn cell accumulations in ipsilateral subventricular zone (SVZ) and peri-infarct regions after stroke. For both hypoperfusion and transient focal cerebral ischemia rats received consecutive labeling of bromodeoxyuridine (BrdU) for 3 days after stroke and examined on day 5. The sections allocated for BrdU staining underwent pretreatment in 2 N HCl at 37 °C for 30 min. Sections were then immunostained with BrdU (1:500) followed by secondary antibodies conjugated to biotin. Then sections were determined using diaminobenzidine (DAB) staining, and positive cells were counted using light microscopy. Five sections of each brain were used to count the BrdU+ cells for peri-infarct and SVZ regions. Representative pictures of BrdU+ cell accumulations in SVZ (a) and peri-infarct (b) regions. Scale bars 100 μm. c DAPT promoted the BrdU+ cell accumulations in peri-infarct and SVZ regions after hypoperfusion. d DAPT promoted the BrdU+ cell accumulations in peri-infarct and SVZ regions after focal cerebral ischemia. Values are mean ± SEM; *P < 0.05, ANOVA, n = 6
DAPT Modulated the Phenotypes of New Born Cells
We checked the phenotypes of these increased new born cells induced by DAPT in penumbra on day 5. Nestin+/BrdU+ colabelings were significantly enhanced (Fig. 6a, d), suggesting increased production of newborn immature neurons and glias in DAPT-treated ischemic penumbra. Though an increased fraction of BrdU+/ laminin+ was not observed in the presence of DAPT, DAPT potentially increased the vascular densities in penumbra on day 14 (Fig. 6c, f). Finally, the fraction of BrdU+/Iba1+ cells was significantly reduced by DAPT in penumbra in comparison with that in vehicle-treated ischemic group (Fig. 6b, e).

DAPT modulated expressions of phenotypic markers after focal ischemia. For transient focal cerebral ischemia rats received consecutive labeling of bromodeoxyuridine (BrdU) for 3 days after stroke and examined in penumbra on day 5 (phenotype analysis) or day 14 (vascular density checking). Sections were immunostained with BrdU (1:500) for dividing cells, glial fibrillary acidic protein (1:500) for mature astrocytes; NG2 (1:300) for oligodendrocytes; nestin (1:500) for immature neurons and glia; NeuN (1:500) for mature neurons; doublecortin (DCX) for migrating neuroblasts; Iba1 (1:500) for microglias; and laminin (1:500) for the vessels, followed by secondary antibodies conjugated to the fluorescent dyes Alexa Fluor 488, 633, and 546, respectively. Cells were checked using a confocal microscope, and cell phenotypes were identified by colocalization of phenotypic markers with BrdU. When possible, at least 100 BrdU+ cells were scored for each marker per animal. For semiquantification of vessels in penumbra, three views in penumbra per section were subjected to the qualification. Four sections of each brain were used to detect the vessels. Representative stainings of BrdU+ co-labeled with Nestin (a) and Iba1 (b), and representative stainings of vessels (c) in penumbra. Scale bars 20 μm. Arrows point on examples of co-labeling cells. d DAPT (gray bar) increases the percentages of BrdU/Nestin co-labeling cells compared vehicle-treated rats (white bar). e DAPT (gray bar) decreased the percentages of BrdU/Iba1 co-labeling cells compared vehicle-treated rats (white bar). f DAPT (gray bar) increased the vascular densities in penumbra at day 14 after stroke compared vehicle-treated rats (white bar). Values are mean ± SEM; *P < 0.05, ANOVA, n = 6
DAPT Regulated the Gene Profiles Associated with Neurogenesis in Penumbra After Focal Cerebral Ischemia
To analyze genes involved in neurogenesis, we employed the Oligo GEArray DNA Microarray Rat Neurogenesis and Neural Stem Cell Gene Array containing 263 known genes (Fig. 1c). Figure S1A shows the profiles of genes in non-stroke, stroke, or DAPT-treated stroke penumbra tissues from representative gene arrays. As expected, housekeeping genes, positive controls show hybridization signals, while blank negative controls show the absence of any hybridization signals. To identify the relevant differentially expressed genes, all clones with more than 1.5-fold estimated differences were considered for further evaluation. This threshold is based on a statistical analysis using online software provided by SuperArray Company.
In non-stroke corresponding penumbral region, 30 out of 263 genes were detected on the neurogenesis and neural stem cell gene arrays (Fig. S1A and Table 1). Many of the 30 genes are associated with neural progenitor cells. These genes included transcription factors, growth factors, and genes involved in signaling pathways and cell cycles.
In ischemic penumbral region, 48 of 263 genes were detected on the gene arrays (Fig. S1A and Table 1). Twenty eight out of 48 genes were the same as detected in non-stroke corresponding penumbral region. S100a6 and Bmp6 were not detected under ischemic conditions. But ischemic penumbra recaptured 20 genes associated with G-protein coupled receptor protein signaling pathway, Wnt receptor/frizzled signaling pathway, cell differentiation, cell adhesion, cell cycle, transcription factor, and growth factor.
In penumbral region under DAPT-treated conditions, 52 of 263 genes were detected on the gene arrays (Fig. S1A and Table 1). Twenty nine out of 52 genes were the same as detected in non-stroke corresponding penumbral region. Bmp6 were not detected as under ischemic conditions without DAPT administration. However, 23 genes were induced under DAPT-treated conditions. In comparison with non DAPT-treated stroke, 16 genes were still detected; 4 genes were not detected including Inha, Hdac4, Cd9, and Efna1; 7 genes were further induced by DAPT. When genes with more than 1.5-fold estimated differences were considered for further evaluation, S100a6 and Cdk5rap3 were markedly upregulated by DAPT after stroke; 7 genes were induced by DAPT including Chrna4, Ncoa6, Ptprz1, Nrp1, Chrm1, Msx1, and Mt3; but 15 genes were markedly downregulated by DAPT after stroke including Cldn11, Gap43, Olig1, Limk1, Drd2, Stmn1, Ywhah, Ache, Acsl6, Adora2a, Ptn, Inha, Hdac4, Cd9, and Efna1. These DAPT-downregulated genes were associated with G-protein coupled receptor protein signaling pathway, cell adhesion, and transcription factor and growth factor.
We performed qRT-PCR analysis for selective seven genes in penumbra (Acsl6, Olig1, Drd2, S100a6, Inha, Hdac4, and Nrp1) (Fig. S1B, C) at day 5 after stroke, and verified the DAPT-induced up/downregulation of genes observed in microarrays.
Discussion
Previous studies have revealed that Notch signaling contributes to neuronal death after ischemia by enhancing apoptotic cascades in neurons in adult brain, thus the treatment with a γ-secretase inhibitor to block Notch signaling pathway may reduce brain ischemic damage [13]. Because the immature brain is vulnerable to hypoxic and ischemic insults, we tested the effects of DAPT against ischemic injury in neonatal rats. We demonstrated that DAPT-induced neuroprotection against neonatal stroke: rescued the ischemic rats from death, attenuated neurons to undergo apoptosis in penumbra, and decreased the infarct size. Neonatal stroke has increasingly been recognized as a significant cause of mortality and long-term neurological deficits in newborns [3]. To ameliorate such outcomes, various experimental treatment modalities, such as hypothermia [4, 5], xenon and sevoflurane inhalation [6, 7], and hypoxic/ischemic preconditioning [8], have been offered. For further improvements in outcome, new treatment strategies, such as pharmacological intervention, therapy combination, and so on, applied to clinic treatment may be beneficial [22]. Our results revealed that DAPT was a robust neuroprotectant against neonatal stroke, suggesting future treatment strategies such as clinical trials using γ-secretase inhibitors would be an attractive therapy for perinatal ischemia.
Although the physiological roles of Notch signaling in neurogenesis and synaptic plasticity have been studied extensively [9–11], its roles played in ischemia-stimulated neurogenesis remain relatively uncertain. Using neural progenitor cells isolated from the SVZ of the adult rat subjected to focal cerebral ischemia, Wang’s group recently investigated the Notch pathway in regulating proliferation and differentiation of these cells after stroke. Blockage of the Notch pathway significantly reduced stroke-induced cell proliferation. Inhibition of the Notch pathway substantially increased neurons, but did not alter astrocytic population in ischemic neural progenitor cells. Their data suggested that the Notch signaling pathway mediated adult SVZ neural progenitor cell proliferation and differentiation after stroke [23]. Another study found that ependymal cell quiescence was actively maintained by Notch signaling. Inhibition of this pathway in uninjured animals allowed ependymal cells to enter the cell cycle and produce olfactory bulb neurons, whereas forced Notch signaling was sufficient to block the ependymal cell response to stroke [24]. Our findings showed that inhibition of Notch signaling in vivo did not reduce the stroke-induced cell proliferation: DAPT increased the newly born cell accumulations in peri-infarct area following ischemia, but inhibition of Notch signaling in vivo promoted the new born cells from 3 days following ischemia to differentiate to immature neurons and glias, which was consistent with those findings mentioned above. Altogether, the Notch signaling pathway fulfills important roles in neonatal ischemia-stimulated neurogenesis. It has been suggested that stroke-induced neurogenesis contributes to neuroprotection and functional recovery after stroke [15–19], thus Notch signaling pathway plays key roles in brain repair may in part through regulating neurogenesis after stroke. However, one study about neonatal stroke in mice found that at first, the majority of cells died in an environment where edema developed; later, over the following weeks, increased apoptosis was visible in the injured hippocampus and neocortex [25]. Another research found that γ-secretase inhibitor treatment decreased infarct size in a reperfusion model of stroke by reducing neuronal apoptosis and decreasing the immune response [13]. So, relieving edema after stroke in the acute period, later, inhibition of neuronal apoptosis and the immune response may also play important roles in brain repair. Notch signaling was activated after stroke [26] with the consequence that the ischemic brain tissues captured multiple detrimental parameters. Therefore, inhibition of Notch signaling by DAPT may suppress these detrimental parameters as well as induce functional molecules via other pathways to modulate tissue repair after stroke. One of our findings supports this speculation that DAPT decreased the fraction of BrdU+/Iba1+ cells in penumbra. Iba1 is a calcium-binding protein and is specifically expressed in microglias in the brain. Stroke insults are accompanied by a marked acute inflammatory reaction, involving the activation of microglias [19]. The majority of evidences to date overwhelmingly suggest a detrimental role for microglial activation [27, 28]. This inhibitory effect on microglial activation by DAPT may in part contribute to the neuroprotection against ischemic injury. We also found that DAPT specifically increased the number of Brdu/Nestin double staining cells at 5 days after stroke; although we did not research function of newborn neuron after stroke, some researchers found that layer 1 neuronal progenitor cells are a source of adult neurogenesis under ischemic conditions in neocortex, and the newly generated neurons were GABAergic and that the neurons were functionally integrated into the neuronal circuitry [29]. So, presumably, part of the newborn neuron has some function. Another finding is that DAPT increases the vascular density in penumbra, which may be due to latter newborn cells adopt this fate because of the inhibition of Notch signaling pathway, and newborn neurons present in the ischemic penumbra surrounding cerebral cortical infarcts in patients with stroke are preferentially localized in the vicinity of blood vessels [30]; our results on angiogenesis are beneficial for the tissue remolding and latter functional recovery after neonatal stroke.
Our findings point to inhibition of Notch signaling elicits some morphologic hallmarks such as neurogenesis and angiogenesis in penumbra that may relate to the brain repair and functional recovery after stroke in the immature brain. We next employed cDNA microarray to analyze gene profiles involved in this regulation of neurogenesis by DAPT. Compared with stroke without DAPT, S100a6 and Cdk5rap3 were markedly upregulated; seven genes were induced. S100a6 is a calcium-binding protein implicated in many cellular processes. Its various biological effects possibly originate from the fact that it may bind to other proteins and modulate their function by inducing conformational changes or interfering with posttranslational modifications [31]. Cdk5rap3 is known to be involved in cell proliferation and differentiation. Among those DAPT-induced genes in ischemic penumbra, Chrna4, Chrm1, and Mt3 were also involved in synaptic functions, such as regulation of synaptic plasticity and synaptic transmission, suggesting an important role for Notch signaling in a form of synaptic plasticity. DAPT, Notch signaling inhibitor facilitating tissue’s repair from ischemic injury, may, at least in part through regulating neurogenesis and synaptic plasticity [32]. These data indicate that penumbra recaptures embryonic molecular signals under DAPT-treated condition after stroke and provide insight into the molecular mechanisms, which regulate the biological function of neural progenitor cells in the penumbra of the brain.
In summary, neonatal stroke results in the long-term neurological deficits. Research found that neonatal ischemia induces a progressive brain injury with prolonged apoptosis and Notch-2 up-regulation. Notch-2 expression was induced shortly after injury and increased cell death. Long-term induction of Notch-2 also occurred in and around areas of cell death; results suggest that neonatal stroke has long-lasting effects on neuronal viability [25]. Ultimately, a large number of neurons died and cause neurological deficits. We have demonstrated that DAPT, a γ-secretase inhibitor, not only rescued the neonatal rats who sustained severe global hypoperfusion or focal cerebral ischemia from death, saved the ischemic brain tissues, but also elicited some morphologic hallmarks such as neurogenesis and angiogenesis in penumbra after stroke that intimately associated with the brain repair and functional recovery. DAPT did regulate the gene profiles associated with neurogenesis in penumbra, which provide information for future molecular and cellular intervention of neurogenesis to facilitate functional recovery after stroke. The present results suggest that DAPT is a potential neuroprotectants against ischemic injury in immature brain. These pharmacological interventions may provide a new direction for treatment strategies to neonatal stroke.
References
Jordan LC, van Beek JG, Gottesman RF, et al. Ischemic stroke in children with critical illness: a poor prognostic sign. Pediatr Neurol. 2007;36(4):244–6.
Brobeck BR, Grant PE. Pediatric stroke: the child is not merely a small adult. Neuroimaging Clin N Am. 2005;15(3):589–607.
Gulati S, Kalra V. Stroke in children. Indian J Pediatr. 2003;70(8):639–48.
Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663–70.
Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574–84.
Thoresen M, Hobbs CE, Wood T, et al. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J Cereb Blood Flow Metab. 2009;29(4):707–14.
Luo Y, Ma D, Ieong E, et al. Xenon and sevoflurane protect against brain injury in a neonatal asphyxia model. Anesthesiology. 2008;109(5):782–9.
Schifrin BS, Ater S. Fetal hypoxic and ischemic injuries. Curr Opin Obstet Gynecol. 2006;18(2):112–22.
Breunig JJ, Silbereis J, Vaccarino FM, et al. Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc Natl Acad Sci U S A. 2007;104(51):20558–63.
Guentchev M, McKay RD. Notch controls proliferation and differentiation of stem cells in a dose-dependent manner. Eur J Neurosci. 2006;23(9):2289–96.
Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442(7104):823–6.
Evin G, Sernee MF, Masters CL. Inhibition of gamma-secretase as a therapeutic intervention for Alzheimer’s disease: prospects, limitations and strategies. CNS Drugs. 2006;20(5):351–72.
Arumugam TV, Chan SL, Jo DG, et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med. 2006;12(6):621–3.
Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445(7129):781–4.
Zhang RL, Zhang ZG, Chopp M. Ischemic stroke and neurogenesis in the subventricular zone. Neuropharmacology. 2008;55(3):345–52.
Teng H, Zhang ZG, Wang L, et al. Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J Cereb Blood Flow Metab. 2008;28(4):764–71.
Abdipranoto A, Wu S, Stayte S, et al. The role of neurogenesis in neurodegenerative diseases and its implications for therapeutic development. CNS Neurol Disord Drug Targets. 2008;7(2):187–210.
Taupin P. Stroke-induced neurogenesis: physiopathology and mechanisms. Curr Neurovasc Res. 2006;3(1):67–72.
Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36(12):2718–24.
Zhao H, Shimohata T, Wang JQ, et al. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25(42):9794–806.
Ling L, Hou Q, Xing S, et al. Exogenous kallikrein enhances neurogenesis and angiogenesis in the subventricular zone and the peri-infarction region and improves neurological function after focal cortical infarction in hypertensive rats. Brain Res. 2008;1206:89–97.
Hobbs CE, Oorschot DE. Neonatal rat hypoxia-ischemia: long-term rescue of striatal neurons and motor skills by combined antioxidant-hypothermia treatment. Brain Pathol. 2008;18(3):443–54.
Wang L, Chopp M, Zhang RL, et al. The Notch pathway mediates expansion of a progenitor pool and neuronal differentiation in adult neural progenitor cells after stroke. Neuroscience. 2009;158(4):1356–63.
Carlen M, Meletis K, Goritz C, et al. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009;12(3):259–67.
Alberi L, Chi Z, Kadam SD, et al. Neonatal stroke in mice causes long-term changes in neuronal Notch-2 expression that may contribute to prolonged injury. Stroke. 2010;41(10 Suppl):S64–71.
Liu XS, Zhang ZG, Zhang RL, et al. Stroke induces gene profile changes associated with neurogenesis and angiogenesis in adult subventricular zone progenitor cells. J Cereb Blood Flow Metab. 2007;27(3):564–74.
Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev. 2006;12(2):105–12.
Biran V, Joly LM, Heron A, et al. Glial activation in white matter following ischemia in the neonatal P7 rat brain. Exp Neurol. 2006;199(1):103–12.
Ohira K, Furuta T, Hioki H, et al. Ischemia-induced neurogenesis of neocortical layer 1 progenitor cells. Nat Neurosci. 2010;13(2):173–U151.
Jin K, Wang X, Xie L, et al. Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A. 2006;103(35):13198–202.
Slomnicki LP, Nawrot B, Lesniak W. S100A6 binds p53 and affects its activity. Int J Biochem Cell Biol. 2009;41(4):784–90.
Wang Y, Chan SL, Miele L, et al. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004;101(25):9458–62.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the National Natural Science Foundation of China (30471840 and 30570644) and by Zhejiang Provincial Natural Science Foundation of China (Y205127 and R2090266).
Conflict of Interest
The authors declare that they have no competing interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 34 kb)
Figure S1
N-[N-(3, 5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) regulated the gene profiles associated with neurogenesis in penumbra at 5 days after focal cerebral ischemia. The Oligo GEArray DNA Microarray Rat Neurogenesis and Neural Stem Cell Gene Array containing 263 known genes were used in the experiments. Microarray protocols were as shown in Fig. 1b. a Representative gene arrays show the profiles of genes in non-stroke, stroke, or DAPT-treated stroke penumbral tissues. All clones with more than 1.5-fold estimated differences were considered for further evaluation. Red and blue circles indicate upregulated and downregulated genes, respectively, detected in microarrays under DAPT-treated conditions compared with genes under vehicle-treated ischemia condition in the penumbra. Real-time RT-PCR analysis of mRNA levels in the penumbra. Some upregulated and downregulated genes (b) detected in microarrays under DAPT-treated condition after stroke compared with levels in vehicle-treated stroke penumbra. Quantification of expression levels of upregulated and downregulated genes (c). Values are mean ± SEM; *P < 0.05, ANOVA, n = 4. (JPG 767 kb)
(JPG 256 kb)
Rights and permissions
About this article

Cite this article
Li, Z., Wang, J., Zhao, C. et al. Acute Blockage of Notch Signaling by DAPT Induces Neuroprotection and Neurogenesis in the Neonatal Rat Brain After Stroke. Transl. Stroke Res. 7, 132–140 (2016). https://doi.org/10.1007/s12975-015-0441-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-015-0441-7