
Abstract
Thrombin mediates the life-threatening cerebral edema and blood–brain barrier (BBB) damage that occurs after intracerebral hemorrhage (ICH). We previously found that the selective cannabinoid receptor 2 (CB2R) agonist JWH-133 reduced brain edema and neurological deficits following germinal matrix hemorrhage (GMH). We explored whether CB2R stimulation ameliorated thrombin-induced brain edema and BBB permeability as well as the possible molecular mechanism involved. A total of 144 Sprague–Dawley (S-D) rats received a thrombin (20 U) injection in the right basal ganglia. JWH-133 (1.5 mg/kg) or SR-144528 (3.0 mg/kg) and vehicle were intraperitoneally (i.p.) injected 1 h after surgery. Brain water content measurement, Evans blue (EB) extravasation, Western blot, and immunofluorescence were used to study the effects of a CB2R agonist 24 h after surgery. The results demonstrated that JWH-133 administration significantly decreased thrombin-induced brain edema and reduced the number of Iba-1-positive microglia. JWH-133 also decreased the number of P44/P42(+)/Iba-1(+) microglia, lowered Evans blue extravasation, and inhibited the elevated matrix metallopeptidase (MMP)-9 and matrix metallopeptidase (MMP)-12 activities. However, a selective CB2R antagonist (SR-144528) reversed these effects. We demonstrated that CB2R stimulation reduced thrombin-induced brain edema and alleviated BBB damage. We also found that matrix metalloproteinase suppression may be partially involved in these processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spontaneous intracerebral hemorrhage (ICH) is a devastating disease. It constitutes 10–15 % of all strokes in the USA, Europe, and Australia and 20–30 % of strokes in Asia. Approximately 2 million cases of ICH are reported annually worldwide [1]. There is currently no effective treatment for ICH, and it has a 1-month mortality rate of 30 to 50 %. Patients who survive typically have major neurological impairments [2].
Cerebral edema is primarily responsible for secondary injury after ICH [3]. Edema increases mass effect and intracranial pressure (ICP) following ICH, which may directly damage brain tissue and ultimately result in herniation [3]. Edema is also directly toxic to neurons and glia by changing osmotic gradients and disrupting the blood–brain barrier (BBB) [4, 5]. Multiple pathways, including cytotoxic injury due to coagulation factors and a robust inflammatory response, lead to edema formation, and thrombin is the primary molecule that mediates the development of acute cerebral edema after ICH [6–8]. The inhibition of thrombin with agratroban or hirudin also reduces edema after ICH in rats [8, 9]. Thrombin infusions into the caudate-putamen of the rat brain induces a rapid increase in edema within several hours that peaks from the first to the third day, and the edema declines gradually over several weeks [10, 11]. The trend of cerebral edema changes in parallel with changes in BBB permeability [11]. Thrombin activates many intracellular signaling cascades in brain cells [12]. P44/42 MAPK, also called extracellular signal-regulated kinase (ERK), is one MAP kinase that is activated in the brain after intracerebral infusions of thrombin [13].
The endocannabinoid system, including endogenous ligands, cannabinoid receptors, and degrading enzymes, is an important pharmacological target in many neurological diseases [14]. Cannabinoid receptor type 1 (CB1R) and cannabinoid receptor type 2 (CB2R) are the most studied cannabinoid receptors [15, 16]. The psychoactive effects of cannabinoids are associated with CB1R, which is predominantly expressed by neurons [17]. The psychoactive effects of CB1R agonists limit their therapeutic potential, which leaves CB2R agonists as the practical option [18]. CB2R is primarily expressed in immune cells, and it mediates anti-inflammatory actions, immune cell migration, cytokine production, and antigen presentation [19]. The anti-inflammatory and neuroprotective effects of cannabinoids in the brain were studied in animal models of multiple sclerosis (MS) and Alzheimer’s disease (AD) [20]. These effects were observed using pharmacological ligands that act on CB1R, CB2R, or both receptors [18]. We previously demonstrated that a specific CB2R agonist (JWH133) attenuated brain edema in rat models of germinal matrix hemorrhage (GMH), but the underlying mechanisms are not known [21].
Increased metalloproteinase (MMP) expression is a key mechanism for increased BBB permeability after ICH [3]. MMPs disrupt BBB integrity and promote edema by degrading tight junction proteins, type IV collagen, laminin, and fibronectin [22, 23].
The present study investigated the effects of a CB2R agonist in a rat model of thrombin-induced BBB damage and the role of MMPs in the neuroprotective process.
Materials and Methods
The selective CB2R agonist JWH133 (Tocris Bioscience) exhibits a very high affinity for the CB2R (Ki = 3.4 nmol/L) but low affinity for the CB1R (Ki = 677 nmol/L). SR144528 (Santa Cruz) is a selective CB2R antagonist. JWH-133 and SR144528 were dissolved in DMSO/ethanol/0.9 % saline (1:1:18) and injected intraperitoneally into each animal. Untreated animals received an equal volume of vehicle (DMSO/ethanol/0.9 % saline (1:1:18)). Thrombin (Sigma) was dissolved in saline at a concentration of 4000 U/ml (163 mg/ml), and thrombin activity was expressed in NIH units.
Animal Preparation and Groups
Adult male Sprague Dawley rats (250–300 g) were housed under specific pathogen-free conditions with free access to food and water until use. Animal use procedures complied with the guide for the care and use of laboratory animals, and the animal care and use committee at the Third Military Medical University approved all procedures. All experiments were designed to minimize the number of animals used and their suffering.
Animals were randomly assigned to the following groups: sham-operated (sham group, n = 36), thrombin + vehicle (vehicle group, n = 36), thrombin + JWH133 (JWH group, n = 36), and thrombin + SR144528 + JWH133 (SR + JWH group, n = 36). All animals were sacrificed 24 h after surgery. All experimental groups and analyses were performed in accordance with RIGOR Guidelines for translational research [24, 25].
Surgery
Model induction was performed as previously reported [26]. Briefly, a feedback-controlled heating pad was used to maintain body temperature at 37.0 °C. Rats were anesthetized with an intraperitoneal injection of chloral hydrate (5 %, 350 mg/kg) and placed in a stereotaxic frame. A cranial burr hole (1 mm) was drilled 4.0 mm lateral to the bregma. Thrombin solution in a volume of 5 μl/rat was micro-infused using a pump at a constant rate of 0.5 μl/min into the right basal ganglia (coordinates: 0.2 mm anterior, 5.5 mm ventral, and 4.0 mm lateral to the bregma) through a 29-gauge needle. The injection needle was left in place for at least 5 additional min to prevent the backflow of drugs. The burr hole was sealed with bone wax, and skin incisions were closed with sutures after the needle was removed. A third group received an intraperitoneal injection of JWH-133 (1.5 mg/kg, JWH group) 1 h after surgery. A fourth group was treated with SR144528 (3 mg/kg) with JWH-133 and 3 min later (1.5 mg/kg) intraperitoneally (SR + JWH group). A second group of animals was injected with an equal volume of vehicle (vehicle group). The sham group only received a needle insertion. The doses of JWH133 and SR144528 were selected based on a previous publication [27].
Brain Water Content Measurement
Brain water content was examined in rats 24 h after surgery, as previously described [26]. Animals (n = 12, per group) were anesthetized with an intraperitoneal injection of chloral hydrate (5 %, 350 mg/kg). Brains were removed, and a coronal tissue was sliced (4 mm thickness) around the injection needle tract. Brain sections were divided into 4 parts: ipsilateral basal ganglia (Ipsi-BG), ipsilateral cortex (Ipsi-CX), contralateral basal ganglia (Cont-BG), and contralateral cortex (Cont-CX). The cerebellum (Cerebel) was the internal control. Brain sample weights were determined immediately after removal and after drying for 24 h in a 100 °C oven using an electric analytical balance. Brain water content (%) was calculated as (wet weight-dry weight)/wet weight × 100 %.
Evans Blue Assay
Evans blue extravasation was performed 24 h post-surgery as previously described [28]. Briefly, Evans blue dye from Sigma-Aldrich (2 %, 4 mL/kg) was injected (>2 min) into the left femoral vein and allowed to circulate for 60 min. Rats under anesthesia (5 % chloral hydrate, 350 mg/kg) (n = 6) were euthanized by an intracardial perfusion with phosphate-buffered solution (PBS), and brains were removed. The right basal ganglia were harvested for homogenization. Samples were weighed, homogenized in saline, and centrifuged at 15,000g for 30 min. An equal volume of trichloroacetic acid was added to the resulting supernatant. Samples were incubated overnight at 4 °C and centrifuged at 15,000g for 30 min. The resulting supernatants were spectrophotometrically quantified at 615 nm for the detection of Evans blue dye extravasation.
Brains for Evans blue fluorescence were removed and fixed in 4 % paraformaldehyde at 4 °C for 24 h. Brains (n = 6) were prepared for coronal brain sectioning (30 μm), and red auto-fluorescence of Evans blue was observed on the slides using a confocal microscope (Zeiss, LSM780) equipped with a 633 nm HeNe laser. A minimum of 4 images were captured for each rat.
Immunofluorescence Staining
Immunofluorescence staining was performed as previously described [27]. The right basal ganglia were infused for 24 h, and rats (n = 6, each group) were re-anesthetized (5 % chloral hydrate, 350 mg/kg) and perfused intracardially with PBS followed by 4 % paraformaldehyde. Brains were removed, post-fixed in 4 % paraformaldehyde for 24 h, and dehydrated in a 30 % sucrose solution for 3–5 days at 4 °C. Free-floating coronal brain slices (30 μm thick) were cut using a cryostat and stored at −20 °C until used. Sections were rinsed with PBS and permeabilized with 0.3 % Triton X-100 in PBS for 30 min. Specimens were blocked with 10 % goat serum for 1 h and incubated at 4 °C overnight with a primary rabbit polyclonal anti-Iba1 antibody (1:200; WAKO Pure Chemical Industries Ltd.) followed by an Alexa 488-labeled goat anti-rabbit IgG (H + L) (1:500, Beyotime, Wuhan, China) for 3 h at 37 °C. A sequential immunofluorescence protocol was used for double immunofluorescence with anti-Iba1 and anti-phospho-p44/42 MAPK antibodies with the appropriate controls. Briefly, free-floating slices were incubated with a primary mouse anti-phospho-p44/42 MAPK (1:200; CST) at 4 °C overnight followed by an Alexa 555-labeled goat anti-mouse IgG (H + L) (1:500; Beyotime, Wuhan, China) secondary antibody (3 h, 37 °C). Sections were washed and blocked with 10 % normal goat serum for 1 h. Sections were incubated overnight with the anti-Iba1 antibody followed by Alexa 488-labeled goat anti-rabbit IgG (H + L) (1:500; Beyotime, Wuhan, China) secondary antibody incubation (3 h, 37 °C). The same protocol was used to investigate the colocalization of ZO-1 and vWF. Sections were permeabilized with 0.3 % Triton X-100 in PBS for 30 min, blocked with 10 % goat serum for 1 h, and incubated at 4 °C overnight with primary antibodies: goat anti–ZO-1 (1:200, Santa Cruz) and mouse anti-vWF (1:200, Santa Cruz). Sections were incubated with appropriate secondary antibodies for 3 h at 37 °C. Colocalization was examined using a fluorescent microscope (Zeiss, LSM780).
Western Blot Analysis
Western blot assays were performed as described previously [29]. Protein extraction of the right basal ganglia tissue (n = 6), including the injection site, was performed 24 h after thrombin injection by tissue homogenization in RIPA buffer (Santa Cruz) supplemented with protease and phosphatase inhibitors (Sigma). Homogenates were centrifuged at 14,000×g at 4 °C for 20 min. Supernatants were whole cell protein extracts and stored at −80 °C until usage. Tissue samples were taken from 6 rats in each group, and one sample was taken from each brain. The protein concentration was determined using a Bio-Rad Laboratories (Hercules, CA, USA) protein assay kit. A total of 50 μg of protein from each sample was loaded into each lane of SDS-PAGE gels. Gel electrophoresis was performed, and proteins were transferred to a nitrocellulose membrane. The membrane was blocked in Carnation® nonfat milk and probed with primary and secondary antibodies. The following primary antibodies were used: anti-phospho-p44/42 MAPK (T202/Y204) (1:1000, CST), anti-p44/42 MAPK (1:1000, CST), antiβ-Tubulin (1:1000, Santa Cruz), anti-MMP-9 (1:1000, CST), anti-MMP-12 (1:1000, Abcam), anti-ZO-1 (1:500, Santa Cruz), and anti–GAPDH (1:1000, Santa Cruz). The membranes were incubated under gentle agitation at 4 °C overnight, and the membranes were washed in TBST. Membranes were incubated in the appropriate HRP-conjugated secondary antibody (diluted 1:1000 in secondary antibody dilution buffer) for 1 h at 37 °C. Protein bands were visualized using a nickel-intensified DAB solution, and the densitometric values were analyzed using ImageJ software. The housekeeping protein β-tubulin and GAPDH were used as internal controls.
Statistical Analysis
Data are reported as the means ± standard derivation (SD). SPSS 13.0 software package (SPSS, Inc., Chicago, IL, USA) was used for statistical analyses. Data were analyzed using one-way analysis of variance (ANOVA) tests followed by Student-Newman-Keuls (SNK) tests. A nonparametric test (Kruskal-Wallis H) was used if the data were not normally distributed, followed by a Nemenyi test when a two-group comparison was necessary. Differences were considered significant at P < 0.05.
Results
Treatment with JWH-133 Decreased Brain Water Content 24 h After Thrombin Infusion
Brain water content of rats in the vehicle group was significantly greater than the sham group 24 h after thrombin infusion, especially in the ipsilateral basal ganglia (Ipsi-BG: sham, 77.45 ± 0.21 % versus Vehicle, 80.24 ± 0.40 %, p < 0.05, Fig. 1). Brain edema in the ipsilateral basal ganglia was significantly reduced 24 h after JWH-133 administration (Ipsi-BG: JWH, 79.32 ± 0.46 % versus vehicle, 80.24 ± 0.40 %, p < 0.05; versus SR + JWH, 80.13 ± 0.46 %, p < 0.05) compared to the vehicle and SR + JWH groups. Brain edema in the ipsilateral cortex (Ipsi-CX) was significantly increased at 24 h (Ipsi-CX: sham, 78.47 ± 0.21 % versus vehicle, 79.35 ± 0.39 %, p < 0.05), and JWH-133 treatment reduced edema levels 24 h post-administration (Ipsi-CX: JWH 78.38 ± 0.40 % versus vehicle, 79.35 ± 0.39 %, p < 0.05; versus SR + JWH, 79.33 ± 0.41 %, p < 0.05) compared with the vehicle and SR + JWH groups (n = 12).

CB2R agonist significantly reduced thrombin-induced brain edema 24 hr after injury. Brain sections (4 mm) were divided into 4 parts: ipsilateral basal ganglia (Ipsi-BG), ipsilateral cortex (Ipsi-CX), contralateral basal ganglia (Cont-BG), and contralateral cortex (Cont-CX). Cerebellum (Cerebel) was the internal control. Values are expressed as the means ± SD, n = 12. Vehicle vs. sham **P < 0.01, vs. JWH # P < 0.05, JWH vs. SR + JWH &P < 0.05
JWH-133 Administration Suppressed Microglial Activation Surrounding the Injury Boundary
Iba1 is an indicator of microglial activation. Iba1 immunoreactivity was revealed using fluorescence microscopy to investigate whether JWH-133 affected microglial activation after surgery (Fig. 2). No obvious microglial activation was expected in the sham group. Many activated microglial cells were widely observed in the vehicle group. The intraperitoneal administration of JWH-133 post-surgery greatly reduced the number of activated microglial cells (Fig. 2a). However, the selective CB2R antagonist SR144528 reversed this treatment effect. Similar results were obtained when the number of Iba-1 positive microglia was quantified (Fig. 2b). Cell number quantification using ImageJ software was performed, as indicated in Fig. 2c, in four pictures of the region surrounding the injury.

Effect of JWH-133 on microglial cell activation surrounding the injection site. Representative images of Iba-1(+) cells at the injection site in sham, vehicle, JWH and SR + JWH groups (a). The number of Iba-1(+) cells around the injection site (b, n = 6 in each group). Select coronal sections of fields of view for immunohistochemical observation (c). Scale bars = 20 μm. Vehicle vs. sham **P < 0.01, vs. JWH # P < 0.05, JWH vs. SR + JWH &P < 0.05
JWH-133 Protects Against Thrombin-Induced Blood–Brain Barrier Destruction
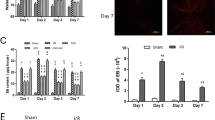
We used Evans blue extravasation to evaluate BBB integrity after surgery. The results demonstrated increased Evans blue dye leakage from vessels within the boundary of the injection site 24 h after surgery, and JWH-133 treatment significantly reduced Evans blue leakage (JWH, 1.70 ± 0.32 versus vehicle, 3.04 ± 0.38, P < 0.05; versus SR + JWH, 2.65 ± 0.34, P < 0.05). Simultaneous SR144528 administrated abolished this effect (Fig. 3b, n = 6). Evans blue immunofluorescence was performed to confirm the extravasation results (Fig. 3a, n = 6), and the same results were obtained.

JWH-133 treatment significantly reduced Evans blue dye leakage around the lesion site. Representative perivascular Evans blue fluorescence around the injection site (a). Scale bars = 20 μm. Assay of extravasation demonstrated that thrombin induced higher Evans blue dye leakage, which can be reduced by a CB2R agonist (JWH133). Moreover, the effect of JWH133 was reversed by SR144528 (b, P < 0.05, n = 6)
JWH-133 Reduces the Phosphorylation Level of p44/42 MAPK After Thrombin Infusion
We examined the phosphorylation level of p44/42 MAPK using Western blot analysis to further clarify the role of the p44/42 MAPK pathway. Western blots demonstrated that the phosphorylation level of p44/42 MAPK markedly increased in the ipsilateral basal ganglia 24 h after the intracerebral infusion of thrombin compared to the sham group (Fig. 4c, P < 0.05, n = 6). However, protein phosphorylation levels were lower in the JWH-133-treated group compared to the vehicle group (P < 0.05). The combination treatment of JWH-133 with the CB2R antagonist SR-144528 reversed protein phosphorylation levels compared to the JWH-133 group (P < 0.05).We performed double immunofluorescence using a combination of antibodies against phosphorylated p44/42 MAPK and cell type-specific protein markers to identify the cell types exhibiting p44/42 MAPK phosphorylation 24 h after thrombin injection. Immunostaining revealed that most of the phosphorylated p44/42 MAPK-positive cells colocalized with the microglial marker Iba1 (Fig. 4a, n = 6). The number of p44/42 MAPK-positive microglial cells decreased after JWH-133 administration 24 h after surgery. SR-144528 administration reversed this effect.

JWH-133 suppressed the phosphorylation level of p44/42 MAPK, and phosphorylation of p44/42 MAPK was primarily visible in reactive microglia. Confocal immunofluorescence images of Iba1 (green), P-ERK (red), and their merged image 24 h after thrombin injection (a). Administration of JWH-133 reduced the phosphorylation of p44/42 MAPK-positive microglia. Phosphorylated p44/42 MAPK, total p44/42 MAPK and β-tubulin proteins in right basal ganglia 24 h after surgery (b). Relative density analyses of phosphorylation levels of p44/42 MAPK (c, p < 0.05, n = 6). Scale bars = 20 μm. Values are expressed as the means ± SD
JWH-133 Prevents Thrombin-Induced ZO-1 Attenuation
We examined BBB integrity using immunofluorescence staining and Western blotting. The tight junction (TJ)-related protein ZO-1 was examined using immunofluorescence microscopy in conjunction with an endothelial marker, von Willebrand factor (vWF), which is also a marker for the BBB. ZO-1 and vWF signals aligned perfectly in the sham group, but this alignment was interrupted while in the vehicle group, which indicates damage to the BBB. However, in the JWH-133 group, some rescue of the superimposed ZO-1 and vWF lining was observed, which suggests an attenuation of the BBB destruction after surgery. However, this effect was abolished with simultaneous administration of SR-144528 (Fig. 5). Notably, Western blot analyses of lysates also demonstrated a significant reduction in ZO-1 levels after surgery, and JWH-133 upregulated the expression of the tight junction protein ZO-1 compared with the vehicle and SR + JWH groups (P < 0.05) (Fig. 6a, d). Immunofluorescence and Western blot analyses demonstrated that JWH-133 treatment obviously protected against TJ protein reduction after injury, which was indicated by the changes in fluorescence and immunoblotting signal intensities in each experimental group. These results further demonstrated that JWH-133 treatment effectively rescued BBB destruction after injury, possibly as a result of attenuated neuroinflammation.

CB2R agonist reduced thrombin-induced BBB damage 24 h after injury. Representative immunohistochemistry staining of ZO-1 (green) and von Willebrand factor (vWF) (red) 24 h after surgery. Arrow indicates the breakdown of continuous endothelial cell layer. Scale bars = 20 μm

Changes in MMP-9, MMP-12, and ZO-1 expression after treatment 24 h post-intracerebral infusion of thrombin. Representative bands (a) and relative density analyses of MMP-9 (b), MMP-12 (c), and ZO-1 (d) expression in the ipsilateral right basal ganglia of brain specimens 24 h after surgery, n = 6. Vehicle vs. sham **P < 0.01, vs. JWH # P < 0.05, JWH vs. SR + JWH, &P < 0.05
JWH-133 Downregulates MMP-9 and MMP-12 Expression After Infusion
MMPs degrade the TJ proteins of the BBB. Therefore, we examined MMP expression in each experimental group. Our results demonstrated that JWH-133 significantly reduced MMP-9 levels in brain tissues near the injury region 24 h post-surgery compared to the vehicle and SR + JWH-133 groups (Fig. 6a, b, P < 0.05). JWH-133 significantly downregulated MMP-12 expression compared to the vehicle and SR + JWH-133 groups (Fig. 6a, c, P < 0.05, n = 6).
Discussion
This study investigated the effects of CB2R activation using a model of intracerebral infusion of 20 U thrombin in rats. Our data demonstrated that the administration of the selective CBR2 agonist JWH-133 after surgery reduced brain water content and Evans blue extravasation.
Our results indicate that suppression of microglial activation and the downregulation of p44/42 MAPK phosphorylation mediated the neuroprotective effects of JWH-133, improved BBB integrity, and restrained MMP-9/12 activity. The CB2R selective antagonist SR-144528 reversed these neuroprotective effects.
Thrombin is a serine protease that is produced immediately after ICH and converts fibrin to fibrinogen to initiate clot formation [3]. Low thrombin concentrations are neuroprotective in vitro and in vivo. A low dose of thrombin attenuated brain edema induced by thrombin or intracerebral hemorrhage and significantly reduced infarct size and brain edema in a rat middle cerebral artery occlusion model via a phenomena called thrombin preconditioning (TPC) [30, 31]. In contrast, high thrombin concentrations are deleterious to the brain after intracerebral hemorrhage [30]. Thrombin is also primarily responsible for early brain edema formation following intracerebral hemorrhage (ICH) [8].
Thrombin is activated through the coagulation cascade once ICH occurs in humans or animal models, and it rapidly diffuses into the brain parenchyma. Therefore, intracerebral thrombin infusion provides a model for thrombin diffusion into the brain after ICH [32]. One milliliter of whole blood produces ~260 to 360 U of thrombin, and a 50-μL clot (used experimentally in rats) produces up to ~15 U of thrombin [6]. Therefore, we injected 20 U of thrombin into the right basal ganglia of the rat to achieve an approximate acute concentration of 35 U/ml of thrombin in the cerebrospinal fluid (CSF) in this study [32] based on an estimated volume of CSF in a 300 g rat of ~580 μl [33]. Activation of the coagulation cascade and production of thrombin disrupts the BBB approximately 24-h post-ICH, which promotes edema formation [6, 8]. Therefore, we chose 24 h after surgery as the time point for our evaluations.
Microglia constitute up to 10 % of the total cell population of the brain, and these cells act as resident macrophages and immune cells of the brain [34, 35]. Microglial activation may contribute to the pathogenesis of brain injury in intracerebral hemorrhage (ICH), and it is also associated with BBB damage. Activated microglia undergo proliferation, chemotaxis, and morphological alterations and generate immunomodulatory molecules [36]. We observed that JWH-133 decreased microglial activation after surgery, as shown by the reduced expression of Iba1 and the predominance of a resting morphology in microglial cells located within the injury boundary. CB2R stimulation inhibits microglia/macrophage cell migration, which may participate in neuroprotection after intracerebral infusion of thrombin. Mitogen-activated protein kinases (MAPKs) are well-known cytoplasmatic signal transducers that play an important role in thrombin-induced neurotoxicity [37]. p44/42 MAPKs are activated in the brain after an intracerebral infusion of thrombin. PD98059 is a specific p44/42 MAPK kinase inhibitor that abolished thrombin-induced activation of p44/42 MAPKs, and it also blocked thrombin-induced brain neurotoxicity [37]. Thrombin treatment also activated p44/42 MAPKs in vitro, and PD98059 completely blocked the cytoprotective effect of thrombin pretreatment, which indicates that the p44/42 MAPK system mediates the thrombin-induced neuroprotective effect [38]. Our study demonstrated that phosphorylation of p44/42 MAPK in reactive microglia was also visible, which may mediate the detrimental effects of thrombin.
ZO-1 anchors the transmembrane protein occludin to the actin cytoskeleton, which confers the capacity of BBB to preclude permeation of blood substances [39]. ICH increases BBB permeability mediated via TJ disruptions with an involvement of MMPs [40]. MMPs are classically known as matrix-degrading enzymes that are involved in many physiological processes, and MMP expression is a key mechanism underlying increased BBB permeability after ICH [41]. Broad-spectrum MMP inhibitors relieve brain injury [42]. An increase in plasma MMP-9 following ICH in humans correlates with peri-hematoma edema and early neurological deterioration. Therefore, MMP-9 is closely associated with edema formation [43, 44]. MMP-12 is not expressed in the healthy brain [45]. MMP-12 is a strong marker of brain injury in animal models. MMP-12 is also the most highly upregulated MMP of the MMPs that were examined after ICH [46]. Microglial activation may release MMPs [47]. Maddahi et al. suggested that inhibition of MEK/ERK signal transduction using a specific raf inhibitor administered up to 6 h after subarachnoid hemorrhage in a rat model normalized the expression of pro-inflammatory mediators and MMP-9 [48]. Adhikary et al. demonstrated that CB2R-selective agonists reduced MMP-9 expression in microglia. Inhibition of MMP-9 is mediated through CB2R-induced reduction in cAMP, inhibition of ERK1/2 AP-1 activation, and the subsequent reduction in AP-1 binding to the MMP-9 promoter [49]. Our Western blot results revealed that JWH-133 suppressed MMP expression after surgery. MMP reduction also correlates with the de-phosphorylation levels of p44/42 MAPK, which may explain why JWH-133 ultimately de-phosphorylates the p44/42 MAPK pathway and suppresses MMPs.
Conclusion
In summary, our data demonstrated that the CB2R agonist JWH-133 attenuated brain edema by preserving BBB integrity following an intracerebral infusion of thrombin. We also found that dephosphorylation of the p44/42 MAPK pathway and the suppression of MMPs such as MMP-9 and MMP-12 were likely involved in the process. These data suggest that C2R agonists are a promising treatment option for BBB protection after ICH.
References
Adeoye O, Broderick JP. Advances in the management of intracerebral hemorrhage. Nat Rev Neurol. 2010;6(11):593–601. doi:10.1038/nrneurol.2010.146.
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–31. doi:10.1016/s1474-4422(12)70104-7.
Bodmer D, Vaughan KA, Zacharia BE, Hickman ZL, Connolly ES. The molecular mechanisms that promote edema after intracerebral hemorrhage. Transl Stroke Res. 2012;3 Suppl 1:52–61. doi:10.1007/s12975-012-0162-0.
Yang GY, Chen SF, Kinouchi H, Chan PH, Weinstein PR. Edema, cation content, and ATPase activity after middle cerebral artery occlusion in rats. Stroke; J Cereb Circ. 1992;23(9):1331–6.
Freeman WD, Barrett KM, Bestic JM, Meschia JF, Broderick DF, Brott TG. Computer-assisted volumetric analysis compared with ABC/2 method for assessing warfarin-related intracranial hemorrhage volumes. Neurocrit Care. 2008;9(3):307–12. doi:10.1007/s12028-008-9089-4.
Hua Y, Keep RF, Hoff JT, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke; J Cereb Circ. 2007;38(2 Suppl):759–62. doi:10.1161/01.STR.0000247868.97078.10.
Xi G, Wagner KR, Keep RF, Hua Y, de Courten-Myers GM, Broderick JP, et al. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke; J Cereb Circ. 1998;29(12):2580–6.
Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996;84(1):91–6. doi:10.3171/jns.1996.84.1.0091.
Kitaoka T, Hua Y, Xi G, Hoff JT, Keep RF. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke; J Cereb Circ. 2002;33(12):3012–8. doi:10.1161/01.str.0000037673.17260.1b.
Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke; J Cereb Circ. 2002;33(10):2478–84.
Guan JX, Sun SG, Cao XB, Chen ZB, Tong ET. Effect of thrombin on blood brain barrier permeability and its mechanism. Chin Med J. 2004;117(11):1677–81.
Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84(1):3–9.
Xi G, Hua Y, Keep RF, Duong HK, Hoff JT. Activation of p44/42 mitogen activated protein kinases in thrombin-induced brain tolerance. Brain Res. 2001;895(1–2):153–9.
Kreitzer FR, Stella N. The therapeutic potential of novel cannabinoid receptors. Pharmacol Ther. 2009;122(2):83–96. doi:10.1016/j.pharmthera.2009.01.005.
Devane WA, Dysarz 3rd FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34(5):605–13.
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–4. doi:10.1038/346561a0.
Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des. 2008;14(23):2347–61.
Ramirez SH, Hasko J, Skuba A, Fan S, Dykstra H, McCormick R, et al. Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood–brain barrier dysfunction under inflammatory conditions. J Neurosci: Off J Soc Neurosci. 2012;32(12):4004–16. doi:10.1523/JNEUROSCI.4628-11.2012.
Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. CB2 receptors in the brain: role in central immune function. Br J Pharmacol. 2008;153(2):240–51. doi:10.1038/sj.bjp.0707584.
Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Exp Rev Mole Med. 2009;11, e3. doi:10.1017/S1462399409000957.
Tao Y, Tang J, Chen Q, Guo J, Li L, Yang L, et al. Cannabinoid CB2 receptor stimulation attenuates brain edema and neurological deficits in a germinal matrix hemorrhage rat model. Brain Res. 2015;1602:127–35. doi:10.1016/j.brainres.2015.01.025.
Romanic AM, Madri JA. Extracellular matrix-degrading proteinases in the nervous system. Brain Pathol. 1994;4(2):145–56.
Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2(7):502–11. doi:10.1038/35081571.
Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4(3):279–85. doi:10.1007/s12975-012-0209-2.
Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490(7419):187–91. doi:10.1038/nature11556.
Jiang Y, Wu J, Hua Y, Keep RF, Xiang J, Hoff JT, et al. Thrombin-receptor activation and thrombin-induced brain tolerance. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2002;22(4):404–10. doi:10.1097/00004647-200204000-00004.
Zarruk JG, Fernandez-Lopez D, Garcia-Yebenes I, Garcia-Gutierrez MS, Vivancos J, Nombela F, et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke; J Cereb Circ. 2012;43(1):211–9. doi:10.1161/STROKEAHA.111.631044.
Chen Y, Zhang Y, Tang J, Liu F, Hu Q, Luo C, et al. Norrin protected blood–brain barrier via frizzled-4/beta-catenin pathway after subarachnoid hemorrhage in rats. Stroke; J Cereb Circ. 2015;46(2):529–36. doi:10.1161/STROKEAHA.114.007265.
Tang JH, Yan FH, Zhou ML, Xu PJ, Zhou J, Fan J. Evaluation of computer-assisted quantitative volumetric analysis for pre-operative resectability assessment of huge hepatocellular carcinoma. Asian Pac J Cancer Prev : APJCP. 2013;14(5):3045–50.
Hua Y, Keep RF, Hoff JT, Xi G. Thrombin preconditioning attenuates brain edema induced by erythrocytes and iron. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2003;23(12):1448–54. doi:10.1097/01.WCB.0000090621.86921.D5.
Xi G, Keep RF, Hua Y, Xiang J, Hoff JT. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke; J Cereb Circ. 1999;30(6):1247–55.
Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, Deng W, et al. Blood–brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010;67(4):526–33. doi:10.1002/ana.21924.
Lai YL, Smith PM, Lamm WJ, Hildebrandt J. Sampling and analysis of cerebrospinal fluid for chronic studies in awake rats. J Appl Physiol Respir Environ Exerc Physiol. 1983;54(6):1754–7.
Thiel A, Heiss WD. Imaging of microglia activation in stroke. Stroke; J Cereb Circ. 2011;42(2):507–12. doi:10.1161/STROKEAHA.110.598821.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi:10.1126/science.1110647.
Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood–brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke; J Cereb Circ. 2006;37(4):1087–93. doi:10.1161/01.STR.0000206281.77178.ac.
Fujimoto S, Katsuki H, Ohnishi M, Takagi M, Kume T, Akaike A. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience. 2007;144(2):694–701. doi:10.1016/j.neuroscience.2006.09.049.
Fujimoto S, Katsuki H, Kume T, Akaike A. Thrombin-induced delayed injury involves multiple and distinct signaling pathways in the cerebral cortex and the striatum in organotypic slice cultures. Neurobiol Dis. 2006;22(1):130–42. doi:10.1016/j.nbd.2005.10.008.
Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869–901. doi:10.1152/physrev.00035.2003.
Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2010;30(4):837–48. doi:10.1038/jcbfm.2009.248.
Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2007;27(4):697–709. doi:10.1038/sj.jcbfm.9600375.
Hamann GF, Burggraf D, Martens HK, Liebetrau M, Jager G, Wunderlich N, et al. Mild to moderate hypothermia prevents microvascular basal lamina antigen loss in experimental focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2004;35(3):764–9. doi:10.1161/01.STR.0000116866.60794.21.
Abilleira S, Montaner J, Molina CA, Monasterio J, Castillo J, Alvarez-Sabin J. Matrix metalloproteinase-9 concentration after spontaneous intracerebral hemorrhage. J Neurosurg. 2003;99(1):65–70. doi:10.3171/jns.2003.99.1.0065.
Castellazzi M, Tamborino C, De Santis G, Garofano F, Lupato A, Ramponi V, et al. Timing of serum active MMP-9 and MMP-2 levels in acute and subacute phases after spontaneous intracerebral hemorrhage. Acta Neurochir Suppl. 2010;106:137–40. doi:10.1007/978-3-211-98811-4_24.
Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12(4):441–5. doi:10.1038/nm1387.
Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Imai Y, et al. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003;53(6):731–42. doi:10.1002/ana.10553.
del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, et al. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2012;32(5):919–32. doi:10.1038/jcbfm.2012.11.
Maddahi A, Ansar S, Chen Q, Edvinsson L. Blockade of the MEK/ERK pathway with a raf inhibitor prevents activation of pro-inflammatory mediators in cerebral arteries and reduction in cerebral blood flow after subarachnoid hemorrhage in a rat model. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2011;31(1):144–54. doi:10.1038/jcbfm.2010.62.
Adhikary S, Kocieda VP, Yen JH, Tuma RF, Ganea D. Signaling through cannabinoid receptor 2 suppresses murine dendritic cell migration by inhibiting matrix metalloproteinase 9 expression. Blood. 2012;120(18):3741–9. doi:10.1182/blood-2012-06-435362.
Acknowledgments
We would like to thank Dr. Ya Hua from the University of Michigan for her professional comments on this research. This work was supported by grants 81571130 (Z.C) and 81070929 (Z.C) from the National Natural Science Foundation of China and 2014CB541606 (H.F) from the National Key Basic Research Development Program (973 Program) of China.
Author Contributions
ZC made substantial contributions to the conception and design. LL and YHT performed the experiments and acquired the data. JT and QWC measured the ventricular volume and cortical length. YJC and YYF participated in tissue fixation and immunohistochemistry. YY and LMY were responsible for supervising all experiments, data analysis and drafting of the manuscript. HF and GZ read and revised some parts of the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
Lin Li, Yihao Tao, Jun Tang, Qianwei Chen, Yang Yang, Zhou Feng, Yujie Chen, Li Ming Yang, Yunfeng Yang, Hua Feng, and Zhi Chen declare that they have no conflicts of interest.
Compliance with Ethics Requirements
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Lin Li and Yihao Tao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Li, L., Tao, Y., Tang, J. et al. A Cannabinoid Receptor 2 Agonist Prevents Thrombin-Induced Blood–Brain Barrier Damage via the Inhibition of Microglial Activation and Matrix Metalloproteinase Expression in Rats. Transl. Stroke Res. 6, 467–477 (2015). https://doi.org/10.1007/s12975-015-0425-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-015-0425-7