
Abstract
The endoplasmic reticulum (ER) functions in the synthesis, folding, modification, and transport of newly synthesized transmembrane and secretory proteins. The ER also has important roles in the storage of intracellular Ca2+ and regulation of Ca2+ homeostasis. The integrity of the Ca2+ homeostasis in the ER lumen is vital for proper folding of proteins. Dysregulation of ER Ca2+ could result in an increase in unfolded or misfolded proteins and ER stress. ER stress triggers activation of the unfolded protein response (UPR), which is a fundamentally adaptive cell response and functions as a cytoprotective mechanism by over-expression of relevant chaperones and the global shutdown of protein synthesis. UPR activation occurs when three key ER membrane-sensor proteins detect an accumulation of aberrant proteins. The UPR acts to alleviate ER stress, but if the stress is too severe or prolonged, apoptosis will be triggered. In this review, we focused on ER stress and the effects of docosahexaenoic acid (DHA) on ER stress. DHA and its bioactive compounds, such as protectins and resolvins, provide neuroprotection against oxidative stress and apoptosis and have the ability to resolve inflammation in neurological diseases. New studies reveal that DHA blocks inositol trisphosphate receptor (IP3R)-mediated ER Ca2+ depletion and ER stress. The administration of DHA post-traumatic brain injury (TBI) reduces ER stress, aberrant protein accumulation, and neurological deficits. Therefore, DHA presents therapeutic potentials for TBI via its pleiotropic effects including inhibition of ER stress.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Endoplasmic reticulum (ER) stress plays a role in the pathogenesis of many neurodegenerative and neurological disorders [1]. The ER is an extensive, interconnected series of membranous sacs that stems from the nuclear envelope and extends throughout the cytosol of all eukaryotic cells. The primary function of the ER is the synthesis, folding, modification, and transport of newly synthesized transmembrane and secretory proteins [2]. Moreover, the ER has important roles in the storage of intracellular Ca2+ and the maintenance of Ca2+ homeostasis within the cell [2]. ER chaperones function to mediate the processes of protein synthesis and protein folding, which requires an optimal ER Ca2+ level [3]. Disruption of Ca2+ homeostasis is associated with a potential increase in unfolded or misfolded proteins, resulting in ER stress. Other causes of ER stress include glucose and oxygen deprivation, an increase in nitric oxide and free radicals, pH shifts, and failure of ER-associated protein degradation (ERAD) [1]. As an attempt to combat ER stress and to ensure some form of protection, the cell evokes the unfolded protein response (UPR) to promote expression of chaperone proteins that aid in protein folding [4]. However, if ER stress is prolonged and there is a sustained activation of the UPR, the cell’s propensity to combat ER stress is exhausted, resulting in the activation of pro-apoptotic pathways, such as the transcription and translation of C/EBP homologous protein (CHOP) and the activation of caspase-12 that subsequently eliminates cells injured by ER stress to ensure the survival of the organism [5, 6].
This review summarizes the role of the UPR and its activation following ER stress, the relationship between ER Ca2+ regulation and ER stress, the activation of the UPR in brain tissues after TBI, and the potential therapeutic effects of DHA on reducing ER stress.
ER Stress and UPR Activation
The UPR is a fundamentally adaptive cell response that, when activated, functions as a cytoprotective mechanism to overcome ER stress by upregulation of relevant chaperones and the global shutdown of protein synthesis [7]. There are three major ER stress sensor-proteins that are associated with the UPR: PKR-like ER kinase (PERK), inositol requiring kinase 1 (IRE1α), and activating transcription factor 6 (ATF6). Activation of the ER stress-sensing pathway results in: (1) the attenuation of protein synthesis that prevents any further accumulation of unfolded proteins, (2) the transcriptional induction of ER chaperone genes to increase the cell’s folding capacity, and (3) the transcriptional induction of the ERAD component genes to aid in the destruction of aberrant proteins [8]. These proteins mediate the alleviation of ER stress with the goal of promoting cell survival with moderate ER stress damage [5].
Under normal physiological conditions when the ER protein folding capacity corresponds to the load of newly synthesized proteins, the activity of these three ER sensor-proteins is suppressed by binding to an ER chaperone, a 78 kDa glucose-regulated protein (GRP78) [9]. However, as conditions of ER stress manifest through the accumulation of misfolded or unfolded proteins in the ER lumen, GRP78 dissociates from the ER stress-sensing proteins, thereby resulting in their activation. Subsequently, GRP78 binds to unfolded proteins to aid in the refolding process [9]. When PERK dissociates from GRP78, PERK oligomerizes and autotransphosphorylates, which leads to its activation. Its kinase domain then phosphorylates the eukaryotic translation initiation factor 2α subunit (eIF2α) that ultimately causes attenuation of global protein synthesis; however, phosphorylation of eIF2α also results in increased translation of specific transcription factors that are upregulated as a result of ER stress, such as ATF3, ATF4, and CHOP [1]. The second ER sensor-protein, IRE1α, has a similar fate as PERK in that it oligomerizes and autotransphosphorylates to its activated form. The activated endonuclease, IRE1α, cuts out a specific 26-nucleotide intron from X-box binding protein 1 (XBP-1) mRNA that is then translated into a transcription factor that targets upregulation of ER stress-related genes, one being GRP78 [10]. Moreover, this pathway results in the activation of the c-Jun N-terminal kinase pathway that activates apoptosis through various mechanisms [1]. ATF6 is the third ER sensor-protein that translocates to the nucleus to promote expression of genes containing the ER stress response element promotor [11]. ATF6-mediated signals serve as a means to counteract ER stress by promoting gene expression of chaperones. These three ER sensor-proteins act to alleviate ER stress, but if the stress is too severe or prolonged, a programmed cell death will be triggered [7]. The activation of the ER membrane associated protein caspase-12 has been linked to ER stress and subsequent programmed cell death. Following activation of the UPR, it is thought that an ER transmembrane protein complex of the chaperone BiP, caspase-7, and caspase-12 dissociates and facilitates the cleavage of caspase-12 creating an apoptotic cascade [1].
ER Ca2+ Dysregulation and ER Stress
As one of the most important intracellular signaling molecules in control of proliferation, differentiation, secretion, contraction, metabolism, trafficking, and cell death, cytosolic Ca2+ is tightly regulated in time, space, and concentration [12]. The ER acts as a dynamic intracellular Ca2+ store and plays an important role in Ca2+ signaling [12, 13]. Ca2+ accumulated within the ER lumen not only controls fast signaling events but also regulates numerous ER-residing chaperone enzymes in post-translational protein processing. These ER chaperones are involved in proper folding of the proteins, and their functional activity is thus tightly regulated by free intraluminal ER Ca2+ concentration [14, 15]. The resting Ca2+ concentration in the ER ([Ca2+]ER) is thousands of times greater than the Ca2+ concentration in the cytosol [16]. This very high [Ca2+]ER acts to drive Ca2+ movement from the ER lumen to the cytosol and controls the concentration and velocity of Ca2+ release which is crucial for cell signaling.
ER Ca2+ homeostasis is precisely regulated by several families of proteins that are responsible for: (1) active Ca2+ accumulation into the ER lumen, (2) Ca2+ storage in the ER, and (3) Ca2+ release in response to appropriate stimulation. Accumulation of Ca2+ ER is accomplished by Ca2+ pumps of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) family [17], which constantly pump Ca2+ against its concentration gradient. The activity of SERCA pumps is controlled by ER Ca2+ content, so that any lowering of [Ca2+]ER immediately increases incoming Ca2+ flux through ER Ca2+ pumps [18, 19]. Ca2+ stored in the lumen of the ER is buffered by Ca2+ binding proteins such as calnexin, calreticulin, calsequestrin, endoplasmin, et cetera. These proteins with a low affinity for Ca2+ (KD ∼ 0.5–1.0 mM) in conjunction with SERCA determine high levels of resting [Ca2+]ER [12, 20]. ER Ca2+ channels are responsible for the controlled Ca2+ release upon appropriate stimulus. The main subtypes of these channels are the ryanodine receptors (RyR) and the inositol triphosphate receptors (IP3R) that are expressed abundantly in most cell types [21, 22]. Under normal physiological conditions, a tight coordination exists between the Ca2+ channels and Ca2+ pumps and thus prevents ER Ca2+ depletion or overload.
In pathological conditions where there is an increased activity of IP3R or RyR, Ca2+ on- and off-mechanisms are compromised that can result in a decreased ER Ca2+ buffering capacity, ER Ca2+ overload, or ER Ca2+ depletion due to the inactivation of SERCA. This will lead to abnormal cytosolic Ca2+ concentration ([Ca2+]cyt) and induce various disease pathologies [23]. As an additional consequence of the perturbed ER Ca2+ homeostasis, post-translational protein processing in the ER is jeopardized and aggregation of aberrant proteins occurs [24]. A disrupted, elevated ER Ca2+ homeostasis can also result in the activation of proteases, phospholipases, and the formation of oxygen- and nitrogen-free radicals [21]. The culmination of a disrupted ER Ca2+ homeostasis and aberrant protein accumulation causes ER stress [5, 25].
ER Stress and Traumatic Brain Injury
The induction of a TBI not only causes direct damage, such as axon shearing, to tissue but also triggers a delayed sequence of cellular and molecular events that result in secondary injury [26, 27]. The mechanisms of sustained secondary injury include molecular events such as disruption of Ca2+ homeostasis, excitotoxicity, and free radical generation [28, 29]. The disruption of Ca2+ homeostasis has been suspected as the fundamental pathological mechanism through which secondary cell injury and death occurs after TBI. TBI is often associated with Ca2+ disruptions and perturbations that could result in the aggregation of unfolded and misfolded proteins. Trauma to the spinal cord has been shown to lead to cell injury and apoptosis due to ER Ca2+ release mediated by both RyR and IP3R [30]. Another study using the fluid percussion injury (FPI) model observed prolonged elevations of hippocampal neuronal Ca2+ levels and a perturbed Ca2+ homeostasis, where affected neurons demonstrated a Ca2+ plateau 1 week post-TBI that returned to baseline levels by 30 days [31]. A disruption of ER Ca2+ homeostasis may result in ER stress and subsequent UPR activation in TBI.
Moreover, direct damage to cellular proteins and folding mechanisms is also associated with TBI and further exacerbates aberrant protein accumulation and thus ER stress [32]. ER stress and the resulting activation of the UPR are involved in acute brain injury after a TBI [1]. In the controlled cortical impact injury (CCI) rat model, increased levels of caspase-12 were observed to peak in the frontal cortex 5 days post-TBI [33]. The expression of CHOP is a hallmark of ER stress and ER-associated programmed cell death [34]. In the CCI model, increased levels of CHOP were observed in the ipsilateral hemispheres of mouse brains 6 h after a sustained TBI and remained elevated 14 days post-TBI [35]. However, it remains unknown whether concurrent activation of caspase-12 and CHOP lead to cell death after TBI. In the FPI model, a decrease in free ubiquitin was observed in the ipsilateral cerebral cortex 7 days post-TBI and in the hippocampus 3 to 7 days post-TBI. Ubiquitin is involved in the regulation of normal cell metabolism and growth and the degradation of aberrant proteins by various ubiquitin ligases [36]. The immunoreactivity of CHOP was also observed in spinal cord tissue at 24 h after the spinal cord injury (SCI) [37]. ER stress-mediated CHOP expression triggers apoptosis and plays a role in neuronal cell death in injured spinal cords [37, 38]. ATF4 and XBP-1, the major transcription factors associated with UPR activation, were also detected at the site of injury 6 h afterward in the SCI model [39]. Moreover, UPR activation was also observed in distant regions from the injured area, indicating that the ER stress response in the SCI model is broad and can have far-reaching deficits in motor-related activities [39]. The mechanisms underlying these broad ER stress responses are unknown; however, pro-inflammatory factors from the lesion site could propagate the events to surrounding tissues [39, 40]. These studies suggest that ER stress contributes to neuronal cell death after TBI. Therapeutic approaches that attenuate ER stress may decrease ER-associated apoptosis and promote neuroprotection. Blocking ER stress is a potential therapeutic option that may also reduce aberrant protein accumulation and promote neuronal recovery after a TBI.
Docosahexaenoic Acid (DHA) and Neuroprotection
DHA is the most abundant omega-3 fatty acid (22:6ω3) found in the central nervous system. It is an essential component of membrane phospholipids with the highest levels found in phosphatidylethanolamine and phosphatidylserine (PS) [41, 42]. Phospholipids containing DHA have been reported to constitute as much as 15–25 mol % of the lipids of the gray matter in the human brain. It is estimated that 50 % of the weight of a neuronal plasma membrane is composed of DHA [43]. In the brain, DHA is involved in development, neurogenesis, memory formation, excitable membrane function, photoreceptor cell biogenesis and function, and neuronal signaling [44].
DHA and Its Effects on Membrane Lipids and Proteins
DHA is the major structural component of the phospholipids in the plasma membrane containing the longest and most unsaturated fatty acid. DHA has been linked to alleviation of some diseases, such as cancers, neurological disorders, cardiovascular diseases, and stroke [45]. The various roles of DHA in a broad range of homeostatic deviations and the mechanisms by which it acts have been a heated question for many years [46]. One possible explanation could be its dynamic effect on membrane properties that in turn modulates intercellular signaling. It is well established that membranes are crucial to the biological process of intercellular signaling. DHA is essential for maintaining the membrane order and fluidity, which is advantageous for neuronal signaling [47, 48]. DHA is capable of undergoing rapid interconversions between multiple torsional states, conferring high permeability, compression, fusion, and flip–flop rates [49]. DHA plays an important role in regulation of PS concentration in the brain, which is essential for cell survival [50]. A lack of DHA reduces the concentration of PS thereby affecting cell signaling and Ca2+ uptake [51]. DHA is a prominent regulator of Ca2+ oscillations within the cell that monitors many cellular functions via either a Ca2+ release or influx to promote gene activation, neurotransmitter release, oxidative stress, or to regulate mitochondrial physiology [51, 52]. These properties make DHA a versatile molecule that can alter the biological properties of cell membranes. In addition, recent studies indicate that DHA may modify protein and lipid organization of the plasma membrane lipid raft structure during essential cell signaling events [41].
DHA can affect the function of multiple targets, ranging from ion channels, nuclear receptors, and second messengers. For example, DHA plays a very important role in maintaining the activity of sodium–potassium pump (Na+/K+-ATPase), a key cell membrane enzyme, that controls ion concentration gradients required for neuronal conduction. The energy derived from the activity of this enzyme accounts for 60 % of energy consumed by the brain [53], which is important in the maintenance of normal physiological processes in the brain. Feeding rats with a DHA-deficient diet lowers the activity of Na+/K+-ATPase [54]. The in vitro activities of protein kinase C, cAMP-dependent protein kinase, mitogen-activated protein kinase (both ERK1 and ERK 2), and Ca2+/calmodulin-dependent protein kinase II (CaMKII) were found to be inhibited by omega-3 fatty acids, such as DHA [55]. In rat olfactory neurons, physiological concentrations of DHA (3–10 μM) are known to inhibit voltage-gated K+ channels and modulate the coding of odorant information by olfactory receptor neurons [56]. DHA also inhibits voltage-sensitive Na+ and Ca2+ channels thereby blocking a depolarization-induced glutamate efflux and subsequent glutamate receptor activation, thus causing a reduction in glutamate-induced excitotoxicity [57]. In the brains of mice treated with a DHA-deficient diet, most of the synaptic proteins that are involved in vesicle trafficking, recycling processes, and neurotransmission were found to be downregulated [58]. The loss of synaptic proteins and the resulting deficit of exocytosis, neurotransmitter release, and vesicle recycling may be associated with a DHA-depletion in the brain [58–61]. Taken together, DHA is essential in maintaining normal neuronal function via the multiple pathways discussed above.
DHA-Mediated Neuroprotection via Anti-inflammation
Oxidative stress and pro-inflammatory responses are components of numerous neuropathological conditions including neurodegenerative diseases (Alzheimer’s disease), amyotrophic lateral sclerosis, spinal cord injury, TBI, epilepsy, and ischemic injury [62, 63]. DHA demonstrated neuroprotective roles against oxidative stress through its ability to scavenge intracellular free radical productions that were induced by hydrogen peroxide, superoxide anions, and hydroxyl radicals in cultured retinal ganglion cells [64–67].
DHA is metabolized into a number of bioactive compounds termed as protectins and resolvins that are capable of not only of resolving inflammation but also providing neuroprotection [68]. Among these bioactive DHA derivatives, neuroprotectin D1 (NPD-1, 10,17S-docosatriene) is an important neuroprotective agent exerting its effects via reducing oxidative stress and tissue inflammation in various neurodegenerative disease conditions, including experimental stroke, Alzheimer’s disease, retinal degeneration, and spinal cord injury [69–74]. NPD-1 limits brain injury associated with stroke by preventing the entry of activated polymorphonuclear leukocytes (PMN) [75]. The neuroprotective mechanisms by which NPD-1 act under conditions of oxidative stress is the upregulation of the anti-apoptotic factors Bcl-2 and Bcl-x(L) and the subsequent downregulation of the pro-apoptotic factors Bax and Bad [76].
Resolvin Ds are formed from DHA through the lipoxygenase (LOX) product 17S-H(p)DHA that is transformed by the LOX activity in human PMN into two epoxide intermediates [75]. The two intermediates then open to form the bioactive products called 17S-resolvin D series (RvD1-4) [75]. Resolvins provide powerful anti-inflammatory and immunomodulatory roles by reducing the migration of neutrophils and the release of pro-inflammatory cytokines [77]. They ultimately protect against damage that is associated with a localized inflammatory response by decreasing the migration of PMN to injured tissues and thereby preventing the oxidative stress that stems from PMN activation [78, 79]. In microglial cells, resolvins block the production of pro-inflammatory cytokines like TNF-α and ILβ1 [80]. DHA also reduces pro-inflammatory mediators such as prostaglandin E2, thromboxanes, and leukotrienes [81].
One of the principal inflammatory signaling pathways affected by DHA is the peroxisome proliferator-activated receptors (PPARs). PPARs are a group of nuclear receptor proteins that function as transcription factors regulating expression of genes [82]. When these receptors are activated, the pathway inhibits the production of pro-inflammatory cytokines that results in a great reduction of inflammation both systemically and in the brain [82]. DHA activates PPARs by mechanisms that remain elusive and in turn suppresses the nuclear factor-kappa b (NF–kb) pathway, a primary mediator of the inflammatory response [83, 84]. In addition, a high dietary intake of DHA reduces inflammation by displacing arachidonic acid, a proinflammatory precursor, and cholesterol from the cell membrane, thereby decreasing the biosynthetic precursors available for production of inflammatory mediators. DHA, through its biosynthetic derivatives resolvins, also inhibits the inflammatory roles of eicosanoids that are produced from arachidonic acid [85]. Taken together, DHA and its biosynthetic derivatives, namely resolvins and protectins, can offer neuroprotection via anti-inflammatory actions and pro-resolving actions.
DHA Reduces ER Stress in In Vitro and In Vivo Studies
In a study by Begum et al. [86], it was shown that DHA has protective effects on astrocyte Ca2+ signaling under oxygen/glucose deprivation and reoxygenation (OGD/REOX) conditions in part by inhibiting Ca2+ dysregulation and ER stress. Two hours of OGD triggered Ca2+ ER store overload (∼1.9-fold), which was then further augmented (∼4.7-fold) at 90 min of REOX [86]. Interestingly, ER Ca2+stores abruptly released Ca2+ at ∼120 min of REOX and then emptied at 160 min of REOX [86]. The depletion of Ca2+ ER stores led to the delayed elevation of intracellular concentrations of cytoplasmic Ca2+ and apoptosis. In contrast, DHA treatment blocked the initial ER Ca2+store overload, as well as the delayed depletion of Ca2+ ER. OGD/REOX-mediated rise in cytoplasmic Ca2+ was significantly attenuated in the presence of DHA, which was in part by inhibiting IP3R [86]. Moreover, exposure of astrocytes to DHA also attenuated the expression of two ER stress markers, p-eIF2α and ATF4. Taken together, these findings suggest that DHA plays a role in reducing ER stress.
In CCI model rats, TBI resulted in a sustained expression of ER stress marker proteins, such as phosphorylated eIF2α, ATF4, and CHOP in the ipsilateral cortex at 3 to 21 days post-TBI [87]. The chronic ER stress was also characterized with an accumulation of abnormal ubiquitin aggregates and an increased expression of the amyloid precursor protein (APP) and phosphorylated tau (p-Tau) in the frontal cortex. The accumulation of APP was colocalized with ER stress marker proteins in the soma. DHA-treated (16 mg/kg in DMSO, i.p.) rats exhibited the attenuation of all ER stress protein expression and a reduced accumulation of both ubiquitinated proteins and APP/p-Tau proteins. The DHA-treated animals also showed early recovery of sensorimotor functions and improved spatial learning and memory 14–20 days post-TBI [88]. It remains to be determined whether post-TBI administration of DHA for 3–7 days significantly reduces ER stress, independent from its protective effects against cell death.
Summary
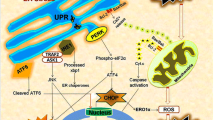
In summary, the ER serves as an important organelle for detecting cellular stress and regulation of cell survival (Fig. 1). Following ischemia or traumatic injury, oxidative stress and a dysregulation of ER Ca2+ could result in an accumulation of unfolded or misfolded proteins and trigger ER stress. ER stress activates the UPR which functions as a cytoprotective mechanism to inhibit global protein synthesis and upregulate key chaperone expression. ER stress, when too severe or prolonged, ultimately stimulates proinflammation and ER stress-associated apoptosis. DHA and its derivatives, protectins and resolvins, provide neuroprotection and resolve inflammation. New studies revealed that DHA also blocks IP3R-mediated ER Ca2+ depletion and ER stress [86]. Post-TBI administration of DHA attenuates ER stress and aberrant protein accumulation and promotes neuronal recovery after TBI. Taken together, in addition to its other pleotropic effects, DHA reduces ER stress and aberrant protein accumulation, which can collectively decrease neurological deficits after brain injury.

ER stress and DHA therapeutic potentials. Ischemia and trauma injuries trigger ER stress via oxidative stress and/or ER Ca2+ dysregulation. ER stress leads to the activation of the PERK, IRE1, and ATF6 pathways that are collectively termed as the unfolded protein response. These pathways result in a global attenuation of protein synthesis, yet an increase in the synthesis of relevant chaperone proteins and ER-associated inflammation. Chronic ER stress and inflammation ultimately lead to apoptosis. The administration of DHA, through its bioactive derivatives, decreases ER Ca2+ dysregulation, can resolve inflammation, and decrease neuronal cell death
Reference
Larner SF, Hayes RL, Wang KK. Unfolded protein response after neurotrauma. J Neurotrauma. 2006;23:807–29.
Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–33.
Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–58.
Buck TM, Wright CM, Brodsky JL. The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:751–61.
Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–83.
DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8.
Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–67.
Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111.
Banhegyi G, Baumeister P, Benedetti A, Dong D, Fu Y, Lee AS, Li J, Mao C, Margittai E, Ni M, Paschen W, Piccirella S, Senesi S, Sitia R, Wang M, Yang W (2007) Endoplasmic reticulum stress. Ann N Y Acad Sci 1113:58-71
Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–59.
Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89.
Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–49.
Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408.
Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium. 2003;34:345–63.
Michalak M, Robert Parker JM, Opas M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 2002;32:269–78.
Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–10.
Wuytack F, Raeymaekers L, Missiaen L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium. 2002;32:279–305.
Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A. Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J. 2002;21:622–30.
Mogami H, Tepikin AV, Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J. 1998;17:435–42.
Papp S, Dziak E, Michalak M, Opas M. Is all of the endoplasmic reticulum created equal? The effects of the heterogeneous distribution of endoplasmic reticulum Ca2+-handling proteins. J Cell Biol. 2003;160:475–9.
Chen X, Kintner DB, Luo J, Baba A, Matsuda T, Sun D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: role of Na+-K+-Cl- cotransporter. J Neurochem. 2008;106:1563–76.
Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–418.
Missiaen L, Robberecht W, van den Bosch L, Callewaert G, Parys JB, Wuytack F, et al. Abnormal intracellular ca(2+)homeostasis and disease. Cell Calcium. 2000;28:1–21.
Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda). 2007;22:193–201.
Paschen W, Hotop S, Aufenberg C. Loading neurons with BAPTA-AM activates xbp1 processing indicative of induction of endoplasmic reticulum stress. Cell Calcium. 2003;33:83–9.
Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–41.
Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12.
Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104.
Sande A, West C. Traumatic brain injury: a review of pathophysiology and management. J Vet Emerg Crit Care (San Antonio). 2010;20:177–90.
Thorell WE, Leibrock LG, Agrawal SK. Role of RyRs and IP3 receptors after traumatic injury to spinal cord white matter. J Neurotrauma. 2002;19:335–42.
Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, et al. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27:1659–72.
Truettner JS, Hu B, Alonso OF, Bramlett HM, Kokame K, Dietrich WD. Subcellular stress response after traumatic brain injury. J Neurotrauma. 2007;24:599–612.
Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90.
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9.
Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, et al. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain Res. 2011;1370:227–37.
Yao X, Liu J, McCabe JT. Ubiquitin and ubiquitin-conjugated protein expression in the rat cerebral cortex and hippocampus following traumatic brain injury (TBI). Brain Res. 2007;1182:116–22.
Wang Z, Zhang C, Hong Z, Chen H, Chen W, Chen G. C/EBP homologous protein (CHOP) mediates neuronal apoptosis in rats with spinal cord injury. Exp Ther Med. 2013;5:107–11.
Chiribau CB, Gaccioli F, Huang CC, Yuan CL, Hatzoglou M. Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2010;30:3722–31.
Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012;3:e272.
Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci U S A. 2011;108:6561–6.
Stillwell W, Wassall SR. Docosahexaenoic acid: membrane properties of a unique fatty acid. Chem Phys Lipids. 2003;126:1–27.
Stillwell W, Shaikh SR, Zerouga M, Siddiqui R, Wassall SR. Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod Nutr Dev. 2005;45:559–79.
Singh M. Essential fatty acids, DHA and human brain. Indian J Pediatr. 2005;72:239–42.
Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–17.
Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood). 2008;233:674–88.
Anderson EJ, Taylor DA. Stressing the heart of the matter: re-thinking the mechanisms underlying therapeutic effects of n-3 polyunsaturated fatty acids. F1000 Med Rep. 2012;4:13.
Suzuki H, Park SJ, Tamura M, Ando S. Effect of the long-term feeding of dietary lipids on the learning ability, fatty acid composition of brain stem phospholipids and synaptic membrane fluidity in adult mice: a comparison of sardine oil diet with palm oil diet. Mech Ageing Dev. 1998;101:119–28.
Ying Z, Feng C, Agrawal R, Zhuang Y, Gomez-Pinilla F. Dietary omega-3 deficiency from gestation increases spinal cord vulnerability to traumatic brain injury-induced damage. PLoS One. 2012;7:e52998.
Hasadsri L, Wang BH, Lee JV, Erdman JW, Llano DA, Barbey AK, Wszalek T, Sharrock MF, Wang H (2013) Omega-3 fatty acids as a putative treatment for traumatic brain injury. J Neurotrauma 30:897-906
Salem Jr N, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–59.
Chalon S, Delion-Vancassel S, Belzung C, Guilloteau D, Leguisquet AM, Besnard JC, et al. Dietary fish oil affects monoaminergic neurotransmission and behavior in rats. J Nutr. 1998;128:2512–9.
Sergeeva M, Strokin M, Reiser G. Regulation of intracellular calcium levels by polyunsaturated fatty acids, arachidonic acid and docosahexaenoic acid, in astrocytes: possible involvement of phospholipase A2. Reprod Nutr Dev. 2005;45:633–46.
Turner N, Else PL, Hulbert AJ. Docosahexaenoic acid (DHA) content of membranes determines molecular activity of the sodium pump: implications for disease states and metabolism. Naturwissenschaften. 2003;90:521–3.
Bourre JM, Francois M, Youyou A, Dumont O, Piciotti M, Pascal G, et al. The effects of dietary alpha-linolenic acid on the composition of nerve membranes, enzymatic activity, amplitude of electrophysiological parameters, resistance to poisons and performance of learning tasks in rats. J Nutr. 1989;119:1880–92.
Mirnikjoo B, Brown SE, Kim HF, Marangell LB, Sweatt JD, Weeber EJ. Protein kinase inhibition by omega-3 fatty acids. J Biol Chem. 2001;276:10888–96.
Seebungkert B, Lynch JW. Effects of polyunsaturated fatty acids on voltage-gated K+ and Na+ channels in rat olfactory receptor neurons. Eur J Neurosci. 2002;16:2085–94.
Vreugdenhil M, Bruehl C, Voskuyl RA, Kang JX, Leaf A, Wadman WJ. Polyunsaturated fatty acids modulate sodium and calcium currents in CA1 neurons. Proc Natl Acad Sci U S A. 1996;93:12559–63.
Sidhu VK, Huang BX, Kim HY. Effects of docosahexaenoic acid on mouse brain synaptic plasma membrane proteome analyzed by mass spectrometry and (16)O/(18)O labeling. J Proteome Res. 2011;10:5472–80.
Yoshida S, Yasuda A, Kawazato H, Sakai K, Shimada T, Takeshita M, et al. Synaptic vesicle ultrastructural changes in the rat hippocampus induced by a combination of alpha-linolenate deficiency and a learning task. J Neurochem. 1997;68:1261–8.
Moriguchi T, Greiner RS, Salem Jr N. Behavioral deficits associated with dietary induction of decreased brain docosahexaenoic acid concentration. J Neurochem. 2000;75:2563–73.
Catalan J, Moriguchi T, Slotnick B, Murthy M, Greiner RS, Salem Jr N. Cognitive deficits in docosahexaenoic acid-deficient rats. Behav Neurosci. 2002;116:1022–31.
Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74.
Adibhatla RM, Hatcher JF. Role of lipids in brain injury and diseases. Futur Lipidol. 2007;2:403–22.
Shimazawa M, Nakajima Y, Mashima Y, Hara H. Docosahexaenoic acid (DHA) has neuroprotective effects against oxidative stress in retinal ganglion cells. Brain Res. 2009;1251:269–75.
Morgane PJ, Austin-LaFrance R, Bronzino J, Tonkiss J, Diaz-Cintra S, Cintra L, et al. Prenatal malnutrition and development of the brain. Neurosci Biobehav Rev. 1993;17:91–128.
Fedorova I, Hussein N, Di MC, Moriguchi T, Hoshiba J, Majchrzak S, et al. An n-3 fatty acid deficient diet affects mouse spatial learning in the Barnes circular maze. Prostaglandins Leukot Essent Fat Acids. 2007;77:269–77.
Kaur P, Heggland I, Aschner M, Syversen T. Docosahexaenoic acid may act as a neuroprotector for methylmercury-induced neurotoxicity in primary neural cell cultures. Neurotoxicology. 2008;29:978–87.
Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A. 2004;101:8491–6.
Bazan NG. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell Mol Neurobiol. 2006;26:901–13.
Bazan NG. Omega-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr Opin Clin Nutr Metab Care. 2007;10:136–41.
King VR, Huang WL, Dyall SC, Curran OE, Priestley JV, Michael-Titus AT. Omega-3 fatty acids improve recovery, whereas omega-6 fatty acids worsen outcome, after spinal cord injury in the adult rat. J Neurosci. 2006;26:4672–80.
Eady TN, Belayev L, Khoutorova L, Atkins KD, Zhang C, Bazan NG. Docosahexaenoic acid signaling modulates cell survival in experimental ischemic stroke penumbra and initiates long-term repair in young and aged rats. PLoS One. 2012;7:e46151.
Eady TN, Khoutorova L, Atkins KD, Bazan NG, Belayev L. Docosahexaenoic acid complexed to human albumin in experimental stroke: neuroprotective efficacy with a wide therapeutic window. Exp Transl Stroke Med. 2012;4:19.
Belayev L, Khoutorova L, Atkins KD, Eady TN, Hong S, Lu Y, et al. Docosahexaenoic acid therapy of experimental ischemic stroke. Transl Stroke Res. 2011;2:33–41.
Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153 Suppl 1:S200–15.
Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–83.
Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat. 2002;68–69:433–55.
Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–22.
Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al. RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. 2006;20:401–3.
Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;278:14677–87.
Litman BJ, Niu SL, Polozova A, Mitchell DC. The role of docosahexaenoic acid containing phospholipids in modulating G protein-coupled signaling pathways: visual transduction. J Mol Neurosci. 2001;16:237–42.
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–41.
Zhao G, Etherton TD, Martin KR, Vanden Heuvel JP, Gillies PJ, West SG, et al. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem Biophys Res Commun. 2005;336:909–17.
Deckelbaum RJ, Worgall TS, Seo T. n-3 fatty acids and gene expression. Am J Clin Nutr. 2006;83:1520S–5.
Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2:355–74.
Begum G, Kintner D, Liu Y, Cramer SW, Sun D. DHA inhibits ER Ca2+ release and ER stress in astrocytes following in vitro ischemia. J Neurochem. 2012;120:622–30.
Begum G, Yan HQ, Shi Y, Zhu W, Dixon CE, Sun D (2012) Docosahexaenoic acid reduces ER stress response after traumatic brain injury in rats. Program No. 769.10. 2012 Neuroscience Meeting Planner. New Orleans, LA; Society for Neuroscience. Online.
Begum G, Yan HQ, Li LL, Singh A, Dixon CE, Sun D (2013) DHA reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. To be submitted.
Acknowledgments
This work was supported in part by the National Institutes of Health Research grant R01NS048216 and R01NS48216 (DS) and the United States Department of Veterans Affairs VA RR&D#B6761R grant (CED).
Conflict of Interest
Gulnaz Begum declares that she has no conflict of interest.
Lloyd Harvey declares that he has no conflict of interest.
C. Edward Dixon declares that he has no conflict of interest.
Dandan Sun declares that she has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Gulnaz Begum and Lloyd Harvey contributed equally.
Rights and permissions
About this article
Cite this article
Begum, G., Harvey, L., Dixon, C.E. et al. ER Stress and Effects of DHA as an ER Stress Inhibitor. Transl. Stroke Res. 4, 635–642 (2013). https://doi.org/10.1007/s12975-013-0282-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-013-0282-1