
Abstract
Traumatic brain injury (TBI) treatment is now focused on the prevention of primary injury and reduction of secondary injury. However, no single effective treatment is available as yet for the mitigation of traumatic brain damage in humans. Both chemical and environmental stresses applied before injury have been shown to induce consequent protection against post-TBI neuronal death. This concept termed “preconditioning” is achieved by exposure to different pre-injury stressors to achieve the induction of “tolerance” to the effect of the TBI. However, the precise mechanisms underlying this “tolerance” phenomenon are not fully understood in TBI, and therefore even less information is available about possible indications in clinical TBI patients. In this review, we will summarize TBI pathophysiology, and discuss existing animal studies demonstrating the efficacy of preconditioning in diffuse and focal type of TBI. We will also review other non-TBI preconditioning studies, including ischemic, environmental, and chemical preconditioning, which maybe relevant to TBI. To date, no clinical studies exist in this field, and we speculate on possible future clinical situations, in which pre-TBI preconditioning could be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A wealth of basic and clinical research exists regarding the treatment of severe traumatic brain injury (TBI). However, despite much research effort, the prognosis for severe TBI patients remains poor [1]. In the USA, an estimated 1.4 million people still suffer a TBI each year [2]. About 50,000 people die before they reach the hospital, and at least 5.3 million live with severe disabilities related to TBI [3, 4]. Worldwide, TBI is recognized as the leading cause of mortality and morbidity in young adults [5]. Globally, thus TBI stands out as a major worldwide health and socioeconomic problem [6, 7].
The most important factor which determinates the prognosis of TBI patients is the severity of the “primary” brain injury. Additional delayed “secondary” brain damage is set in progress, and continues from the time of traumatic impact in TBI patients, and the two combine to determine outcome. Primary brain injury itself is mostly not amenable to treatment; consequently, the strategy of primary TBI treatment should be prevention, such as use of helmets and vehicle modification, etc. Preconditioning strategies offer the theoretical possibility of increasing “resilience” to primary TBI, and also reduction of delayed secondary damage.
The concept of preconditioning has been gaining interest in the field of treatment for both neuronal and cardiac disorders. The term “preconditioning” was first introduced by Janoff in 1964 to describe a noxious stimulus below the threshold of cell injury which results in subsequent protection [8]. Ischemic preconditioning (IPC), also known as “induced tolerance”, refers to the ability of a brief and sublethal ischemic episode, followed by a period of reperfusion, to increase the organ’s resistance to injury following a subsequent ischemic insult [9, 10]. The quote from Friedrich Nietzche, “What which does not kill me makes me stronger” describes the essence of preconditioning.
Initially investigated for cardiac protection, the preconditioning concept has now been extended to several other organs. Current literature in the neurological arena focuses predominately on preconditioning for stroke, but new attractive areas of research are traumatic brain injury, especially subjects involved in violent activities or sports and high-risk elective neurosurgical patients, such as aneurysm reconstructions.
Literature identifies two windows of preconditioning. Immediate preconditioning occurs rapidly (within 1 h after the preconditioning stimulus); it is characterized by potent, but transient changes in cellular enzyme activity, secondary messengers and ion channels. Delayed preconditioning, in contrast, develops slowly, is long-lasting, and involves new gene expression and de novo protein synthesis [11, 12].
Several different types of preconditioning have been described: cross (when the preconditioning stimulus is different from the subsequent noxious stimulus), remote (when preconditioning of one organ then produce protection in a different remote organ), immunological (when activation of a signaling cascade is achieved by pharmacological or immunological compounds), mimetic (when preconditioning is promoted by a compound which mimics the pathways to be blocked), and effector (when the downstream mediators are actually used to prevent an anticipated harmful cellular event) [13].
Many stimuli, such as sublethal ischemia, hypoxia, pharmacological agents, hypothermia, biological agents, anesthetics, seizure, and essentially anything that causes cellular stress, can promote a preconditioning response in the brain. The challenge is whether or not these preconditioning stimuli can then result in effective neuroprotective strategies for clinical use, in the TBI setting. In the field of TBI, preconditioning investigations have been very limited, so far, although results from preconditioning studies in other brain injury areas, e.g., ischemia, have been very promising.
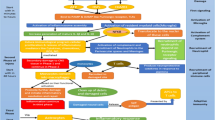
Brain injury pathomechanisms are possibly amenable to preconditioning. The brain injured is heterogeneous, both for pathology and severity of brain injury, and multiple pre-existing comorbidities may coexist in the TBI patient (e.g., ischemic heart disease). Thus, the complexity of TBI pathophysiology (as illustrated in Fig. 1) provides a strong rationale for the use of combination therapies, not only for neuroprotective strategies, but also in the setting of preconditioning, acting at different levels of the secondary injury cascade. It should also be remembered that the number of preconditioning stimuli, the types of preconditioning, as well as the elapsed time between preconditioning and the test insult may affect results [10]. Below, we summarize TBI pathophysiology and discuss the efficacy of preconditioning for each type of TBI (Fig. 1).

Schematic representation of the effect of preconditioning in traumatic brain injury (TBI). In the upper part of the illustration, the main pathomechanisms responsible for the injurious cascade after TBI are represented, which can result in different kind of brain damage and adversely affect neurologic outcome. In the lower part of the figure, preconditioning strategies applied before TBI, acting alone or in combination, result in the phenomenon of preconditioning, which may reduce the extent of subsequent brain damage and result in improved neurologic outcome
Pathomechanisms in TBI Amenable to Preconditioning—Diffuse and Focal TBI
There have been many studies on the pathomechanical classification of TBI [14–17]. TBI can be divided into two components, primary and secondary brain injury
Primary brain injury that occurs at the moment of impact is characterized by diffuse axonal injury (DAI), contusions, and disruption of cranial nerves and vessels. Secondary brain injury will not be as severe immediately after the time of impact; however, it will usually worsen after a primary brain injury in severe TBI with coma. Secondary damage includes mechanisms, such as neuroinflammation, glutamatergic excitotoxicity, oxidative stress, DNA damage, and neuronal cell death by apoptosis and necrosis.
Both primary and secondary brain injury can be further classified according to focal or diffuse mechanisms, although focal and diffuse pathological process are often intermingled, making it difficult to divide into focal, diffuse, primary, and secondary categories, it is useful to consider them separately for the purpose of understanding pathophysiology.
Diffuse Axonal Injury
The best example of diffuse injury is DAI. DAI was first described by Strich [18] as a clinical, pathological syndrome in patients who were unconscious from the time of trauma, with microscopic traumatic axonal damage involving many large white matter tracts, and without intraparenchymal lesions [19]. In DAI, traumatic lesions most commonly affect white matter in areas, such as brain stem, corpus callosum, basal ganglia, thalamus, and the cerebral hemispheres.
DAI is now recognized to typically involve a more progressive response involving a transient, traumatically induced disruption of the axonal membrane over 24–48 h in humans, primarily caused by uncontrolled calcium influx [20] at ion channels near the nodes of Ranvier of axons (Fig. 2) [21].

Microscopic characteristics in DAI. a Schematic illustration which shows the fate of the axon subjected to focal cytoskeletal perturbation [21]. At the site of injury (A) traumatically induced neurofilamentous misalignment detectable after a brief period of survival and depicted in this enlargement (B) results in focal impairment of axonal transport. The subsequent accumulation of organelles results in formation of a reactive axonal swelling, its continued expansion, and its eventual disconnection (C) from the distal segment of the axon, often by 6 h. b Microscopic image of retraction balls (silver stain ×800). This is the most representative, microscopic feature of DAI
The cytoskeletal components of axons include three main proteins: microtubules, neurofilaments, and microfilaments. After suffering traumatic brain injury, calcium-related activation of the calpain system results in proteolysis of the cytoskeletal structure and may play an integral role in delayed neuronal degeneration-calpain-mediated spectrin proteolysis [22]. In axonal distortion injury, within minutes there is malalignment and distortion of the cytoskeletal components [23], which leads to loss of microtubules and increased spacing of neurofilaments, especially at the node of Ranvier. These mechanisms induce a subsequent failure of axoplasmic transportation, pooling of intra-axonal contents, and disconnection of the axon from its distal part. This disconnection occurs over 24 to 72 h after the traumatic impact and is termed delayed or secondary axotomy. The primary impact thus causes axonotomy by secondary biochemical processes, and this phenomenon makes the so called “retraction ball”, the characteristic signature of DAI on microscopy (see also Fig. 2). These delayed pathophysiologic processes suggest that DAI might be also characterized as a secondary brain injury.
At the molecular level, trauma inducing calcium influx initiates calpain activation [24–27] and mitochondrial injury/swelling [28] with cytochrome c release and caspase activation (Fig. 3) [29]. This worsens axonal injury, and may also cause neuronal death by apoptosis [30].

Neuromembrane events in posttraumatic phase. Neurotransmitter release causes the opening of ion channels and influx of Na+ and Ca++. The resultant combination of intracellular volume and Ca2+ overload induces cell swelling, plasma membrane swelling, necrosis, and apoptosis and leads to the activation of destructive enzymes [112]
The glutamatergic system is also an important factor for the development of early necrosis-mediated damage in TBI. This system mainly induces uncontrollable excitotoxicity with the increase of the influx of Ca2+ through N-methyl-d-aspartate (NMDA) receptors [31]. Blocking of excitotoxicity using NMDA receptors antagonists has been included as a neuroprotective strategy in many human and animal studies [32].
Preconditioning for Diffuse Brain Injury
In some experimental studies, diffuse injury animal models were used to test the effect of preconditioning in TBI. A recent experimental study showed that preconditioning with subtoxic NMDA administration was able to protect against novel recognition memory impairment and increase cellular survival in the hippocampus of mice in a closed-head weightdrop model [33], which is universally used for mimicking diffuse injury [34]. They concluded that a sublethal dose of NMDA might modulate the downstream effect of calpain activation [35]. Furthermore, these authors advocated that the pre-activation of NMDA receptors with NMDA administration inhibited neuronal excitability and reduced the influx of Ca2+ at the time of TBI [36].
The effect of heat stress preconditioning in DAI has also been examined. In 1994, Shohami et al. demonstrated that when DAI was induced in heat preconditioned rats, they displayed faster and better functional recovery as well as reduced secondary tissue damage [37]. Heat stress preconditioned rats also showed significantly better motor function 48 h following injury as compared with normothermic controls [37]. Additionally, brain edema and blood–brain barrier disruption were reduced in the acclimated group. These findings were subsequently reinforced in mice, demonstrating significantly better motor function 24-h post-injury accompanied with a reduction in brain water accumulation [38].
In 2008, Su et al. also clarified that heat stress preconditioning had a potential for attenuation of cognitive impairment in the rat model of DAI which was generated by the impact-acceleration method [39]. Moreover, they concluded this effect of heat stress preconditioning might be mediated by the neuroprotective effect of heat shock protein (HSP) 70, which was induced after heat stress and protects cytoskeletal proteins from enzymolytic degradation by caspases [39].
Recent studies have also shown the efficacy of preconditioning with hyperbaric oxygen (HBO). Hu et al. investigated the effect of HBO preconditioning on the weight drop TBI model of DAI. They concluded HBO preconditioning could attenuate TBI in rats, with improvement of cerebral blood flow and brain tissue oxygenation also [40].
Pathophysiology of Focal Brain Injury
Focal brain injuries are usually generated by tissue strains at sites of contact of the brain against the cranial coverings, particularly the more confining ridges of the anterior and middle cranial fossa against the anterior and inferior surfaces of the frontal and temporal lobes. This type of injury very frequently occurs in combination with diffuse brain injury, when the rapidly decelerating brain is displaced within the skull.
Focal Cortical Contusion—a Universal Type of Focal Brain Injury
Focal cortical contusion is usually caused by shearing forces which injure the blood vessels (arteries, veins, and large capillaries) and other structures of the parenchyma (neural cells and glial cells). Contusions often progress within hours to days with evolving events related to the interplay of hemorrhage, vasogenic edema, and ischemic necrosis. In the first 24 h, contused brain tissue biopsies show an inflammatory response, which is predominantly intravascular and consists of vascular margination of polymorphonuclear leukocytes. Extravascular polymorphonuclear leukocytes can be demonstrated in injured brain tissue only a few minutes after TBI. Within 3–5 days, the inflammation is also present in parenchyma and consists of reactive microglia, monocyte-macrophages, polymorphonuclear cells, CD8, and CD4 T-cells [41] generating further neuronal swelling, by complement activation, and other mechanisms. Inflammatory cells also produce free radicals and cytokines, such as tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β), a mediator of blood–brain barrier injury that leads to brain swelling and induces DNA fragmentation in oligodendrocytes [42] and neurons.
Several reports have also provided evidence that, in addition to necrosis, apoptosis also occurs with human cerebral contusions [30, 43]. Focal contusions are also associated with increased metabolic activity in the surrounding tissues. In a rat fluid percussion model study, increase in glucose metabolism occurs immediately after TBI, and is maximally localized to regions of the brain that are maximally deformed by the impact of injury [44]. One study suggested that anaerobic glycolysis occurred as a result of both increased ionic pumping, and excessive glutamate release [45]. These series of reactions exhaust glucose in the extracellular space.
Furthermore, focal cerebral contusion also generates surrounding “ischemic injury”. Histologically, a cerebral contusion has a characteristic necrotic core and a peri-lesional penumbral zone, in which the neurons demonstrate “ischemic changes” such as pyknosis, and vacuolation. In addition, any insult below the threshold to induce necrosis may progress to apoptosis [46]. Therefore, “preconditioning therapy” might be directed towards anti-apoptotic strategies for mild to moderate ischemic damage and anti-necrotic strategies for more severe and prolonged ischemic insults. Blockade of apoptosis may reverse the harmful effects, seen in the peri-lesional area, after contusion TBI.
Preconditioning for Cerebral Contusion
Previous rat studies have applied the concept of preconditioning to a focal cerebral contusion model.
In 1999, the group of Perez-Pinzon and Dietrich described the efficacy of ischemic preconditioning in a moderate fluid percussion brain injury rat model, which generated a focal contusion [9]. They compared CA3 necrotic cell counts and contusion volumes between with and without preconditioning groups. They clarified that ischemic preconditioning applied 48 h before fluid percussion brain injury reduced necrotic cell counts and contusion volumes when compared to the control group [9]. They suggested that ischemia is also implicated in the pathophysiology of some types of TBI which includes focal cortical contusion; therefore, ischemic preconditioning also might be effective in this type of patient.
More recently, Longhi et al. reported the long-lasting neuroprotection by endotoxin preconditioning in a controlled cortical impact brain injury rat model [47]. As preconditioning, a sublethal dose of lipopolysaccharide (LPS) was administrated, and then focal cortical impact brain injury was induced in mice. Mice receiving LPS before TBI showed attenuated motor deficits at 1 week after injury. LPS preconditioning reduced the expression of CD68 and increased IL-6 in injured mice. With these results, the authors concluded that LPS preconditioning might be associated with a modulation of inflammatory responses like microglia/macrophage activity and cytokine production, for example [47].
Subdural Hematoma as the Ischemic/Reperfusional Injury
The reported mortality of subdural hematoma is from 30 to 90 % and worse outcome has been correlated with pathological findings of ischemic brain damage in the hemisphere underlying the hematoma. One of the important causes of ischemia is raised intracranial pressure (ICP), which produces hemispheric cerebral hypoperfusion. Raising ICP reduces cerebral blood flow, especially under the subdural. Removal of the subdural hematoma may thus cause immediate recovery from the global cerebral hypoperfusion, and consequently preventing further ischemic injury. However, sudden reperfusion may also lead to secondary injury after abrupt reduction of a mass lesion [48–50]. Thus, removal of subdural hematoma may be associated with ischemic/reperfusion (I/R) injury which in turn can be reduced by pre-treatments, such as hypothermia or free radical scavengers [51].
As mentioned above, recovery from ischemia may induce an I/R injury. The processes leading to cellular damage after I/R injury are complex and multi-factorial [52] (Fig. 4). At this point the pathology of I/R injury has been separated into two distinct mechanisms. One is the cell death following cellular dysfunction, i.e., excitotoxcity, acidotoxicity, and ionic imbalance. This first process is seen primarily, very early in the ischemic phase. A second type of early I/R injury comes from free radical production, and this becomes particularly disastrous during the reperfusion phase [53]. Together these mechanisms create a complicated picture of injury.

Schematic illustrating the mechanisms of ischemic/reperfusional (I/R) injury and the effects of therapeutic hypothermia. The pathology of I/R injury is approximately separated as two mechanisms, that is, the cell death after cellular dysfunction in ischemic phase, and the free radical production in reperfusion phase. The boxed arrow with entered “Hypothermia” means the estimated effective points in I/R cascade (illustration from [53])
In the early ischemic phase, brain ischemia initiates a cascade of destructive and often irreversible processes that destroy brain cells and tissue. One example of this is the intracellular conversion to anaerobic metabolism due to mitochondrial failure [54]. Depletion of adenosine triphosphate (ATP) in the absence of oxidative metabolism leads to failure of the Na+/K+ ATPase pump (See Fig. 3). This causes depolarization of the cell membrane leading to activation of voltage-gated calcium channels and an influx of intracellular calcium [55]. Moreover, with the anaerobic metabolism induced, intracellular and extracellular acidosis worsens the calcium influx. This rapid increase in intracellular calcium causes release of large amounts of the excitatory neurotransmitter glutamate, which further stimulates calcium influx in post-synaptic cells—a “viscous cycle” [56]. Calcium entry triggers activation of phospholipase, nitric oxide synthase, proteases, endonucleases, and oxidase enzymes [57]. These activated molecules can easily damage other cell proteins and lipid membranes causing necrosis [58]. These processes have been shown to be present within minutes to hours after the injury and probably peak at between 4–6 h [55, 59]. Recent studies have also demonstrated the production of superoxide radicals by NMDA receptor-mediated nicotinamide adenine dinucleotide phosphate oxidase activation [60]. Such events amplify reactive oxygen species (ROS) production, mitochondrial dysfunction, and proapoptotic protein activation. Intracellular calcium accumulation itself also triggers initiation of mitochondrial dysfunction and fragmentation leading to activation of proapoptotic proteins such as the caspases and cytochrome C [61].
Reperfusion of this ischemic tissue results in a short period of excessive free radical production [62]. Experimental measurements in this reperfusion phase demonstrate that oxygen- and carbon-centered free radicals peak within 5 min of reperfusion [63] and that hydroxyl generation peaks within 15 min [64]. This oxidative stress can damage proteins, lipids, and DNA, possibly leading to necrosis and apoptosis [65, 66]. Oxidants also modulate neuro-inflammation [59] leading to increased levels of neuronal apoptosis in adjacent cells [67–69].
Preconditioning for Ischemic/Reperfusion Injury
Exercise preconditioning has been shown to provide endogenous neuroprotection, preserving neuronal viability in the setting of ischemia/reperfusion injury, and resulting in decreased infarct volume and improved neurologic recovery [70–73].
From these previous studies, endogenous neuroprotection induced by exercise preconditioning has been shown to take place through multiple mechanisms, including upregulation of neurotrophin expression, strengthening of the blood–brain barrier, enhancing the cerebral capillary and arterial networks, decreasing inflammation and apoptosis, and improving cerebral metabolism.
Exercise preconditioning also decreases the inflammatory response and leukocyte invasion following ischemia/reperfusion injury, thus decreasing much of the secondary damage seen following the reperfusion stage [74]. Interestingly, “exercise preconditioning” is a universal requirement, for professional contact sports (e.g., NFL football, boxing) and before military combat.
Therapeutic hypothermia also has been shown in previous studies to improve histopathological and behavioral consequences of TBI using many experimental models [75–81], and different delivery paradigms. Hypothermia also has been researched as a preconditioning procedure in ischemic/reperfusion TBI models. The treatment/prevention effect of hypothermia has been reported for several biochemical points in the I/R cascade (Fig. 4).
Our group showed the efficacy of early-induced hypothermia as a preconditioning strategy against reperfusion injury after surgical decompression in acute subdural hematoma (ASDH). We induced ASDH with autologous blood injection into the rat subdural space, and hypothermia (33 °C) was induced 30 min before decompressive craniotomy (DC) in an early hypothermia treatment group. Intercellular ubiquitin carboxyl-terminal hydrolase-L1 (UCH-L1) and glial fibrillary acidic protein (GFAP) were measured with the microdialysis (MD) technique to determine the amount of neuronal and glial damage and the effect of hypothermic preconditioning before surgical decompression was compared to late-induced hypothermia which was induced 30 min after DC as a conventional treatment method. In the normothermia (37 °C) and late-induced hypothermia groups, neuronal and glial reperfusional damage were seen to be severe in the post decompressive phase. On the other hand, pre-reperfusional hypothermic preconditioning mitigated the reperfusional neural and glial damage in the early hypothermia group (Fig. 5).

Efficacy of hypothermic preconditioning for reperfusional injury in the decompressive craniotomy ASDH rat model. Ubiquitin carboxyl-terminal hydrolase-L1 (UCH-L1) and GFAP concentrations in microdialysate. In early phase of reperfusion UCH-L1 MD concentration in normothermia group was highest and significantly higher than sham rat group (*p < 0.01). Also, UCH-L1 MD in early hypothermia group was significantly lower than normothermia group (#p < 0.01). GFAP concentration in normothermia was highest and peaked in the late phase of reperfusion (**p < 0.05 vs sham). Early-hypo early hypothermia group, Late-hypo late hypothermia group, Normo normothermia group
In cerebral and cardiac ischemia, many preconditioning paradigms has been studied and their efficacy has been discussed [82–86]. Unfortunately, as compared to cerebral/cardiac ischemia, there are fewer preconditioning studies in TBI. Also, there are still no clinical trials. However, some parts of the pathophysiology after TBI overlap with the pathophysiology of ischemic disease. Indeed, several types of preconditioning mitigate progressive neuronal damage in I/R TBI. These areas of overlap warrant further research in TBI.
New Studies and Future Directions in Preconditioning Methodology for TBI
NMDA Preconditioning
It is well known that excitatory neurotransmission, through the excessive activation of the N-methyl-d-aspartate receptors, plays a critical role among mechanisms of brain damage, after TBI, and in addition, glutamate, the main excitatory amino acid in the mammalian central nervous system is markedly elevated in the extracellular fluid after TBI. Glutaminergic excitoxicity is responsible of both necrotic and apoptotic death, following primary and secondary injury. Preconditioning with low dose of NMDA has thus been proposed as a neuroprotective strategy in TBI. Recent studies [87] in experimental models of diffuse TBI reported a protective effect of sublethal doses of NMDA in improving motor behavioral deficits in mice, thus supporting the idea that mechanisms of neural death can be limited by NMDA preconditioning, by low level activation of the glutaminergic system through a low dose of NMDA. Furthermore, in another study aiming at investigating cellular viability, memory and other behavioral parameters in an experimental model of mild TBI, it was reported that NMDA preconditioning could protect against neuronal damage in the hippocampus [33]. Furthermore, it was observed that the NMDA did not cause damage per se and protected against both cellular damage and against memory deficits in a novel objective recognition task [33].
It is interesting to note that previous studies on models of brain ischemia demonstrated that IPC promoted a shift from excitatory to inhibitory neurotransmission [88], and this leads to speculation that gamma-aminobutyric acid (GABA) could play a role in promoting ischemic tolerance. To clarify this, further, an analysis of glutamate and GABA concentration by microdialysis was performed. The study revealed that extracellular glutamate concentration was 61 % lower in the ischemic preconditioning group compared with the ischemia group during the first 10 min of reperfusion [88]. Furthermore, in the experiment evaluating if GABA release was increased in the IPC group, a robust significant increase in the extracellular GABA levels was observed in the preconditioned animals (3,112 % compared with the sham group and 510 % compared with the ischemia group). The results of this study suggests that besides glutamate, higher GABA levels after ischemia contribute to lower excitotoxicity in preconditioned animals, and a shift between excitatory and inhibitory neurotransmission may play a critical role in the mechanism of protection by ischemic preconditioning [88].
The role of GABA synapses as a therapeutic target is also suggested by the extensive literature to support the use of antiepileptic drugs targeting postsynaptic GABA receptors. The role of functional modification of GABA synapses and of activation of the ε isoform of protein kinase C (εPKC) in the mechanism of ischemic preconditioning has been recently addressed. In an in vitro study using the technique of miniature postsynaptic current analysis (mPSC) to evaluate neuroprotective changes in GABA synapse function after preconditioning, a significant increase in the number of synapses or release sites was documented, and a significant increase in the amplitude of GABAA mPSCs was seen [89]. Furthermore, inhibition of εPKC abolished the changes in GABA synapses, suggesting that the impact of ischemic preconditioning on GABA synapses depended on downstream effectors and targets of the εPKC signaling pathway [89].
A possible role for glucagon in the protective preconditioning response by impeding the accumulation of glutamate has been recently addressed [90]. Authors proposed that glucagon may be of value in providing neuroprotection when administered after TBI or prior to certain neurosurgical or cardiac interventions in which the incidence of perioperative ischemia is high [90].
Heat Stress Preconditioning
Among physical means of preconditioning, heat stress and exercise are very attractive in the field of neurotrauma. This is especially relevant for the possible clinical implication in specific populations at risk of TBI, such as soldiers and sport combatants, (boxers, fighters, and NFL football) or people involved in any high-risk physical activities.
It has been observed that neurons subjected to heat stress produce heat shock proteins and that once these are produced, neurons can be resistant to different stressors. The high inducible HSP70 is particularly important in the central nervous system. In a study investigating the effect of heat stress preconditioning on learning and memory performances in a model of diffuse axonal injury, HSDAI rats (rats subjected to heat stress preconditioning 24 h before induction of DAI) demonstrated superior spatial learning compared to the DAI alone group [39]. Furthermore, it was observed that memory retention was altered by DAI, protected by heat stress preconditioning and not affected by heat stress alone [39]. In the cerebral cortex and in the hippocampus, there were less axonal retraction balls in the HSDAI rats compared to the DAI group. Preconditioned animals also presented higher expression of HSP70. The results of this study demonstrated that heat stress preconditioning was able to protect against secondary impairment of cognitive function induced after DAI, together with reduced histopathological damage [39].
Previous animal models of closed head injury had demonstrated a role for the transcriptional activator hypoxia-inducible factor 1 (HIF-1), regulating the expression of tens of target genes, in the mechanism of heat acclimation (HA) [38]. Erythropoietin gene is one of the genes regulated by HIF-1. At 4 h after injury, a significant higher level of nuclear HIF-1 and higher levels of EpoR in HA mice were observed as compared with controls and this was related to better clinical recovery, suggesting evidence for neuroprotection [38].
Other experimental observations after TBI also further confirm the ability of HA to reduce the activation of apoptotic pathways in mice [91]. The expression levels of Bad and Bcl-xL, key proteins in the intrinsic apoptosis pathway, were measured, as well as caspase-3 activity. To express the balance between the antiapoptotic Bcl-xL and proapoptotic Bad within the mitochondria, its ratio was calculated, which was higher in HA mice, suggesting the presence of a balance favoring antiapoptosis after heat acclimation [91]. Caspase-3 enzyme activity in normothermic animals was twofold higher than in HA mice. The above results favor attenuation of postinjury cell death, by heat acclimation which affects factors associated with the intrinsic pathways of apoptosis induction, with an effect sustained for up to 8 days after injury [91].
Lipopolysaccharide Preconditioning
Neuroinflammation plays a main role in the pathomechanisms of secondary brain damage, after TBI. It acts via activation of cytokine and complement cascades [92], contributing to development of brain edema and apoptotic cell death. Secondary hypoxic insult, which may occur after the injury, may also upregulate production of inflammatory mediators such as IL-6, IL-1β, and TNFα in an experimental model of diffuse axonal injury [93].
Studies with experimental models of ischemia suggest that toll-like receptors are involved in the enhancement of cell damage following ischemia, and cerebral preconditioning downregulates pro-inflammatory TLR signaling, so this could reduce the inflammation which exacerbates ischemic brain injury [94].
Targeting TLR signaling may be a novel therapeutic strategy for cerebral ischemic injury and other inflammatory diseases, and stimulation of some TLRs prior to ischemia can provide neuroprotection by inducing a state of tolerance to subsequent ischemic injury [94]. Suppression of the normal inflammatory responses to ischemia is a hallmark of the lipopolysaccharide preconditioned brain.
If these concepts can be translated to TBI research, it is evident that preconditioning, acting through a modulation of inflammatory response, can be effective in promoting neuroprotection. In particular, the possibility of modulating microglia/macrophage responses to TBI, by mean of endotoxin preconditioning, has been recently investigated in a model of controlled cortical impact brain injury [47]. It was demonstrated that mice receiving LPS at different times before TBI had a significantly better performance and a significantly smaller contusion volume, when compared to the injured animals. Interestingly, when analyzing mRNA expression of genes known to be modulated by acute brain injury, it was observed that LPS attenuated the posttraumatic increase in CD11b and CD68, and increased that of IL-6, when compared with saline. Preconditioning effects were robust and persisted for up to 1 month after injury [47]. The possible role of TNFα in this effect need to be investigated in future studies.
Based on the above results, induction of immunologic tolerance, or preconditioning through cytokine administration, could play a role, modulating inflammatory cascades following TBI, and could be proposed for populations at risk for brain injury, such as contact sports, soldiers, or high-risk neurosurgical procedures, such as aneurysm reconstruction.
Mitochondrial Preconditioning
Since dysfunctional mitochondria are implicated in acute and chronic neurodegeneration, it is now increasingly recognized that mitochondrial targeted preconditioning may represent a promising therapeutic weapon against neurodegeneration [10–12, 95].
It has recently been proposed that transient exposure of mitochondria to physiopathological intracellular events, or pharmacological agents, determines mitochondrial changes which can protect neurons against adverse subsequent critical events [11]. Mitochondrial respiratory chain, reactive oxygen species, mitochondrial ATP-sensitive potassium channels (mitoKATP), and mitochondrial permeability transition pore (mPTP) are all both targets and mediators of mitochondria-mediated preconditioning [11].
Increasing evidence to support the involvement of mitochondrial ROS and mitoKATP channels in the neuroprotective mechanisms triggered by preconditioning is building. This suggests a fundamental role of mitochondria in the preconditioning phenomenon. While it is well recognized that exaggerated mitochondrial ROS production is associated with mitochondrial dysfunction, neurodegeneration, and death, it is increasingly observed that a slight rise of mitochondrial ROS levels can induce brain tolerance by preconditioning [11–13]. Ravati et al. [96] demonstrated that preconditioning by moderate ROS stimulation protects cultured neurons against different damaging agents and prevents against subsequent massive oxygen radical formation.
It has also been shown that preconditioning can be prevented with the pharmacological inhibition of mitoKATP channels and can be activated by mitoKATP agonists. ROS may directly activate mitoKATP channels [97]. Activation of mitoKATP channels with pharmacological agents thus probably mimics the protective effects mediated by preconditioning and, by limiting Ca2+ overload, prevents mPTP induction. Diazoxide, a selective mitoKATP channel opener, has been proposed as a potent inducer of preconditioning-mediated protection due to the combined effects of mitochondrial membrane depolarization and enhanced ROS production [98].
A key signaling pathway in the protection of brain mitochondria in preconditioning is protein kinase C. Among all PKC isozymes, in particular, the PKC isozyme epsilon (εPKC) is a fundamental signaling pathway after ischemic preconditioning [95]. A role for mitochondrial uncoupling proteins as part of neuroprotective responses involved in preconditioning has also been proposed [11].
Another key transcription factor involved in preconditioning-induced brain tolerance is the hypoxia-inducible factor 1 [12]. In the case of hypoxia, HIF-1α proteasomal degradation is inhibited, resulting in HIF-1α stabilization and translocation to the nucleus. In the nucleus, HIF-1α recruits HIF-1β and modulates the expression of several target genes involved in angiogenesis, metabolism, apoptosis, and cell survival [11, 12]. Recent evidence also support a role for MicroRNA (small RNAs that function as regulators of post-transcriptional gene expression) as mediators and regulator of new protein synthesis in the brain’s response to ischemic preconditioning [99].
The subject of mitochondrial targets for ischemic preconditioning has been recently reviewed by Perez-Pinzon et al. [95]. In their comprehensive review, a detailed synthesis of mitochondrial targets for ischemic preconditioning is provided—ischemic preconditioning activates the adenosine A1 receptor, which then activates phospholipase C (PLC). Phosphatidyl inositol bis-phosphate is hydrolyzed by PLC forming diacylglycerol, a potent activator of protein kinase C isoforms, such as εPKC. εPKC, then, activates the ATP-sensitive mitochondrial potassium channel. Transmembrane cycling of K+ may slightly depolarize the mitochondrial inner membrane and decrease production of ROS. Ischemic preconditioning also alters NAD+/NADH levels, activating the sirtuin 1 (SIRT1) pathway, thus leading to a decrease of the uncoupling protein 2 [95].
Whether or not mitochondrial preconditioning can be proposed as an effective neuroprotective strategy in the prevention of secondary ischemic injury after TBI needs to be extensively evaluated in the experimental and clinical setting.
Anesthetic Preconditioning
Because of their physiological effect on neuronal transmission, antagonism of excitatory neurotransmission, and potentiation of inhibitory neurotransmission, anesthetics have been considered logical candidates for neuroprotection and have been evaluated more than any other drug, class in the field of preconditioning, in ischemia.
Although specific studies on the role of anesthetic preconditioning in TBI are lacking, anesthetic drugs are widely used in clinical practice and can be easily tested both in the experimental and clinical setting for their putative preconditioning effect, in TBI models.
The possibility that administration of volatile anesthetic agents can produce myocardial preconditioning against myocardial ischemic injury was the first to be documented [100], and is still undergoing investigations [101].
Preconditioning and protection against ischemia–reperfusion in non-cardiac organs is also important to understand, especially in the brain. Preconditioning of the brain with pharmacologic agents against ischemic injury might be an attractive, fairly easily implemented strategy to reduce morbidity and mortality from ischemic brain damage especially during high-risk neurosurgical procedures, such as difficult large aneurysm, reconstruction, large tumors, vascular reconstruction, or spinal cord injury. This area deserves further investigation, especially if we consider that volatile anesthetics, because of their known cerebrovasodilation effect, are usually avoided in patients with compromised cerebral hemodynamics, increased intracranial pressure, and impending ischemia, as seen in severe TBI.
Proposed mechanisms of preconditioning with volatile anesthetics in the brain include induction of nitric oxide production, activation of adenosine triphosphate-sensitive potassium channels, and increases in intracellular Ca2++ concentration in the brain. In a preconditioning model in which organotypic cultures of rat hippocampus were exposed to isoflurane for a 2-h period 24 h before an ischemia-like injury of oxygen–glucose deprivation, it was observed that isoflurane preconditions neurons in hippocampal slice cultures by mechanisms that seem to involve intracellular Ca2++ and the MAP kinase–ERK pathway. Preconditioning was also associated with increased levels of the antiapoptotic protein kinase B [102].
The neuroprotective effect of isoflurane is long lasting, being evident even 24 h after administration, and is inducible nitric oxide synthase (iNOS) mediated [103]. The downstream mediators through which iNOS-derived nitric oxide (NO) exerts its preconditioning effect have been recently investigated, with evidence that peroxynitrite, formed from iNOS-derived NO and nox2-derived superoxide, is a key factor in the mechanisms of the tolerance[104].
Other investigations documented in vitro that xenon and sevoflurane preconditioning reduces the amount of neuronal cell death provoked by oxygen–glucose deprivation injury and increased cellular viability through an antinecrotic mechanism in a PI3K-dependent manner [105]. In vivo, the combination of xenon and sevoflurane provided long-lasting neuroprotection, as measured by histologic and neurologic outcomes. Thus, volatile anesthetic preconditioning induces new genes and proteins, via intracellular signals and mediators, such as protein kinases and transcription factors, and PI3K is important also for volatile anesthetic postconditioning [105]. Whether this can only apply to ameliorate the consequences of perinatal and adult hypoxic–ischemic injury, or can also be extended to larger populations, such as severe TBI, need to be further investigated.
Hyperbaric Oxygen Preconditioning
Hyperbaric oxygen therapy can produce ischemic tolerance by preconditioning [106]. In an experimental study investigating the effects of high altitude on traumatic TBI and examining the neuroprotection provided by HBO preconditioning, it was observed that HBO pre-conditioning can attenuate TBI in rats at high altitude by improvement of regional cerebral blood flow and brain tissue oxygenation [107].
Interestingly, in a study investigating the role of HBO in a mouse surgical brain injury model (partial brain resection), it was observed that HBO preconditioning provided protection against SBI by improving neurological outcomes and reducing postoperative brain edema[108]. Furthermore, HBO preconditioning attenuated COX-2 increase after SBI, probably via neuronal suppression of HIF-1a. Less COX-2 could result in lessened brain injury via reduced inflammation, oxidative cell death and apoptosis [108].
Exercise Preconditioning
Physical and mental stressors are putative preconditioning stimuli in the arena of traumatic injury, with several important implications for “high TBI risk” activities, e.g., military combat and in aggressive contact sports, such as boxing, wrestling, and football.
Other than affecting other recognized risk factors such as body weight, blood pressure, serum cholesterol, and glucose intolerance, it has been increasingly observed that exercise per se can play an effective role in inducing endogenous neuroprotection directly against brain damage. This effect has been related to several mechanisms and, among these, an increased production of mRNA for neurotrophins (brain-derived neurotrophic factor-BDNF- and nerve growth factor-NGF-) and increased angiogenesis [97]. Exercised ischemic rats demonstrated a highly significant 79 % reduction in infarct volume as compared with ischemic rats without exercise [73]. A significant increase in microvessel density and cellular expression of NGF and BDNF in cortex and striatum was detected in rats subjected to transient middle cerebral artery occlusion and preconditioned by treadmill exercise for 3 weeks prior to stroke [73].
Although the putative neuroprotective role of exercise has not been fully investigated in experimental models of TBI, nevertheless, some interesting observations related to cerebral metabolic effects of exercise can be translated to the field of brain injury. A recent study [109] evaluated the effect of two different exercise regimens (forced versus voluntary exercise) on glycolytic proteins which play a significant role in cerebral metabolism (glucose transporter 1 (GLUT-1), glucose transporter 3 (GLUT-3), phosphofructokinase (PFK), lactate dehydrogenase (LDH), and 5-AMP-activated protein kinase (AMPK) [109]. In this study, it was observed that the forced exercise group had a significant increase in cerebral glycolysis, including expressions of GLUT-1, GLUT-3, PFK, LDH, phosphorylated AMPK activity, and HIF-1α, when compared to the voluntary exercise and the control groups [109]. The authors demonstrated that the two exercise regimens resulted in increased cerebral glycolysis in connection with HIF-1α upregulation, and this was more evident in the forced exercise regimen [109]. It was suggested that when hypoxia is combined with exercise, the resultant oxidative stress may play an important role in increasing HIF-1 expression. It was also observed that exercise with a stressful component is neuroprotective, while stress alone is not, suggesting a beneficial combination of physical activity in conjunction with induced stress [109].
Other studies in rats subjected to ischemia and reperfusion, demonstrated a significant upregulation of HSP70 and pERK 1/2 after 3 weeks of exercise, and a significant reduction in neuronal apoptosis and brain infarct volume [110]. As observed also from heat stress preconditioning studies, this protein over-expressed in neurons may reduce neuronal apoptosis by upregulating the levels of several anti-apoptotic proteins and downregulating pro-apoptotic proteins [110].
Interestingly, this positive preconditioning effect is long lasting and neuroprotection continued, even after 3 weeks of rest [111]. The multiple putative effects through which exercise preconditioning may affect brain homeostasis have been recently reviewed by Dornbos et al. [74], and involve enhanced integrity of the neurovascular units, “strengthening” of the blood–brain barrier, angiogenesis, decreased inflammatory response, reduced neuronal apoptosis, increased metabolic capacity, upregulation of cerebral blood flow, glucose transport, glycolysis, and ATP production. All these mechanisms should be evaluated in the specific field of neurotrauma, for the possible enormous clinical implications.
Clinical Implications
Since the time of the Spartans (∼800 BC), human history has shown that small groups exercise and stress—“preconditioned” fighters have been much more effective than larger armies. It is tempting to speculate that modern boxing and other contact sportsmen derive neuroprotective benefit from their training regimes, which involve exercise, heat, and stress.
New biochemical detection approaches, such as serum biomarker measurement, may possibly shed light on this hypothesis. Whether our society is ready to specifically precondition volunteers who place themselves at high risk of TBI (such as soldiers and boxers) is unclear. The fact that, in the USA, helmet use for motorcyclists is voluntary in 20 states, suggests otherwise.
Conclusion
To reduce the impact of primary and secondary brain damage, the concept of preconditioning is highly attractive. Several different types of preconditioning have been described, such as cross, remote, immunological, pharmacological, anesthetic, mimetics, and effectors in many type of TBI models. The most robust preconditioning effect has been seen for ischemic pathophysiology. The ischemic mechanism also surely exists as the part of pathomechanisms in TBI, therefore, preconditioning strategies are clearly worth considering for further research in TBI.
References
Langlois JA, Marr A, Mitchko J, Johnson RL. Tracking the silent epidemic and educating the public: CDC’s traumatic brain injury-associated activities under the TBI Act of 1996 and the Children’s Health Act of 2000. J Head Trauma Rehabil. 2005;20(3):196–204.
Binder S, Corrigan JD, Langlois JA. The public health approach to traumatic brain injury: an overview of CDC’s research and programs. J Head Trauma Rehabil. 2005;20(3):189–95.
Fu ES, Tummala RP. Neuroprotection in brain and spinal cord trauma. Curr Opin Anaesthesiol. 2005;18(2):181–7. doi:10.1097/01.aco.0000162838.56344.88.
Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6(1):28–42. doi:10.1016/j.nurt.2008.10.036.
Cole TB. Global road safety crisis remedy sought: 1.2 million killed, 50 million injured annually. JAMA. 2004;291(21):2531–2. doi:10.1001/jama.291.21.2531.
Mar J, Arrospide A, Begiristain JM, Larranaga I, Elosegui E, Oliva-Moreno J. The impact of acquired brain damage in terms of epidemiology, economics and loss in quality of life. BMC Neurol. 2011;11:46. doi:10.1186/1471-2377-11-46.
Clifton GL, Valadka A, Zygun D, Coffey CS, Drever P, Fourwinds S, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 2011;10(2):131–9. doi:10.1016/s1474-4422(10)70300-8.
Janoff A. Alterations in lysosomes (intracellular enzymes) during shock; effects of preconditioning (tolerance) and protective drugs. Int Anesthesiol Clin. 1964;2:251–69.
Perez-Pinzon MA, Alonso O, Kraydieh S, Dietrich WD. Induction of tolerance against traumatic brain injury by ischemic preconditioning. Neuroreport. 1999;10(14):2951–4.
Perez-Pinzon MA. Neuroprotective effects of ischemic preconditioning in brain mitochondria following cerebral ischemia. J Bioenerg Biomembr. 2004;36(4):323–7. doi:10.1023/B:JOBB.0000041762.47544.ff.
Correia SC, Carvalho C, Cardoso S, Santos RX, Santos MS, Oliveira CR et al. Mitochondrial preconditioning: a potential neuroprotective strategy. Front Aging Neurosci, 2010; 2. doi:10.3389/fnagi.2010.00138.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Mitochondria: the missing link between preconditioning and neuroprotection. J Alzheimers Dis. 2010;20 Suppl 2:S475–85. doi:10.3233/jad-2010-100669.
Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. doi:10.1016/s1474-4422(09)70054-7.
Lindenberg R, Freytag E. The mechanism of cerebral contusions. A pathologic-anatomic study. Arch Pathol. 1960;69:440–69.
Maloney AF, Whatmore WJ. Clinical and pathological observations in fatal head injuries. A 5-year survey of 173 cases. Br J Surg. 1969;56(1):23–31.
Freytag E. Autopsy findings in head injuries from firearms. Statistical evaluation of 254 cases. Arch Pathol. 1963;76:215–25.
Adams JH, Graham DI, Scott G, Parker LS, Doyle D. Brain damage in fatal non-missile head injury. J Clin Pathol. 1980;33(12):1132–45.
Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19(3):163–85.
Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11 Suppl 1:5–11.
Pettus EH, Christman CW, Giebel ML, Povlishock JT. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11(5):507–22.
Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12(4):555–64.
Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14(3):121–34.
Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14(7):419–40.
Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58(4):365–75.
Shields DC, Schaecher KE, Hogan EL, Banik NL. Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res. 2000;61(2):146–50.
Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7(1):31–42. doi:10.1016/j.nurt.2009.11.002.
Kilinc D, Gallo G, Barbee KA. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp Neurol. 2009;219(2):553–61. doi:10.1016/j.expneurol.2009.07.014.
Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19(4):443–51. doi:10.1097/00004647-199904000-00010.
Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20(8):2825–34.
Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–22.
Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1(8):623–34.
Willis C, Lybrand S, Bellamy N. Excitatory amino acid inhibitors for traumatic brain injury. Cochrane Database Syst Rev, 2004; (1):CD003986. doi:10.1002/14651858.CD003986.pub2.
Moojen VK, Damiani-Neves M, Bavaresco DV, Pescador BB, Comim CM, Quevedo J, et al. NMDA preconditioning prevents object recognition memory impairment and increases brain viability in mice exposed to traumatic brain injury. Brain Res. 2012;1466:82–90. doi:10.1016/j.brainres.2012.05.041.
Adelson PD, Robichaud P, Hamilton RL, Kochanek PM. A model of diffuse traumatic brain injury in the immature rat. J Neurosurg. 1996;85(5):877–84. doi:10.3171/jns.1996.85.5.0877.
Costa T, Constantino LC, Mendonca BP, Pereira JG, Herculano B, Tasca CI, et al. N-methyl-d-aspartate preconditioning improves short-term motor deficits outcome after mild traumatic brain injury in mice. J Neurosci Res. 2010;88(6):1329–37. doi:10.1002/jnr.22300.
Boeck CR, Kroth EH, Bronzatto MJ, Vendite D. Effect of the L- or D-aspartate on ecto-5’nucleotidase activity and on cellular viability in cultured neurons: participation of the adenosine A(2A) receptors. Amino Acids. 2007;33(3):439–44. doi:10.1007/s00726-006-0455-2.
Shohami E, Novikov M, Horowitz M. Long term exposure to heat reduces edema formation after closed head injury in the rat. Acta Neurochir Suppl (Wien). 1994;60:443–5.
Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E. Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. J Cereb Blood Flow Metab. 2005;25(11):1456–65. doi:10.1038/sj.jcbfm.9600142.
Su Z, Han D, Sun B, Qiu J, Li Y, Li M, et al. Heat stress preconditioning improves cognitive outcome after diffuse axonal injury in rats. J Neurotrauma. 2009;26(10):1695–706. doi:10.1089/neu.2008.0519.
Hu SL, Hu R, Li F, Liu Z, Xia YZ, Cui GY, et al. Hyperbaric oxygen preconditioning protects against traumatic brain injury at high altitude. Acta Neurochir Suppl. 2008;105:191–6.
Clausen F, Lorant T, Lewen A, Hillered L. T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma. 2007;24(8):1295–307. doi:10.1089/neu.2006.0258.
Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci. 2009;14:3795–813.
Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17(10):927–38.
Kawamata T, Katayama Y, Hovda DA, Yoshino A, Becker DP. Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acids. Brain Res. 1995;674(2):196–204.
Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91(22):10625–9.
Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92(16):7162–6.
Longhi L, Gesuete R, Perego C, Ortolano F, Sacchi N, Villa P, et al. Long-lasting protection in brain trauma by endotoxin preconditioning. J Cereb Blood Flow Metab. 2011;31(9):1919–29. doi:10.1038/jcbfm.2011.42.
Miller JD, Bullock R, Graham DI, Chen MH, Teasdale GM. Ischemic brain damage in a model of acute subdural hematoma. Neurosurgery. 1990;27(3):433–9.
Kuroda Y, Bullock R. Local cerebral blood flow mapping before and after removal of acute subdural hematoma in the rat. Neurosurgery. 1992;30(5):687–91.
Burger R, Bendszus M, Vince GH, Solymosi L, Roosen K. Neurophysiological monitoring, magnetic resonance imaging, and histological assays confirm the beneficial effects of moderate hypothermia after epidural focal mass lesion development in rodents. Neurosurgery. 2004;54(3):701–11. discussion 11–2.
Kuroda Y, Fujisawa H, Strebel S, Graham DI, Bullock R. Effect of neuroprotective N-methyl-d-aspartate antagonists on increased intracranial pressure: studies in the rat acute subdural hematoma model. Neurosurgery. 1994;35(1):106–12.
Yokobori S, Frantzen J, Bullock R, Gajavelli S, Burks S, Bramlett H, et al. The use of hypothermia therapy in traumatic ischemic/reperfusional brain injury: review of the literatures. Therapeutic Hypothermia And Temperature Management. 2011;1(4):185–92. doi:10.1089/ther.2011.0012.
Lampe JW, Becker LB. State of the art in therapeutic hypothermia. Annu Rev Med. 2011;62:79–93. doi:10.1146/annurev-med-052009-150512.
Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37(7 Suppl):S186–202. doi:10.1097/CCM.0b013e3181aa5241.
Badruddin A, Taqi MA, Abraham MG, Dani D, Zaidat OO. Neurocritical care of a reperfused brain. Curr Neurol Neurosci Rep. 2011;11(1):104–10. doi:10.1007/s11910-010-0156-9.
Simon RP. Acidotoxicity trumps excitotoxicity in ischemic brain. Arch Neurol. 2006;63(10):1368–71. doi:10.1001/archneur.63.10.1368.
Wahlgren NG, Ahmed N. Neuroprotection in cerebral ischaemia: facts and fancies—the need for new approaches. Cerebrovasc Dis. 2004;17 Suppl 1:153–66. doi:10.1159/000074808.
Leker RR, Shohami E. Cerebral ischemia and trauma—different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Brain Res Rev. 2002;39(1):55–73.
Wong CH, Crack PJ. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr Med Chem. 2008;15(1):1–14.
Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12(7):857–63. doi:10.1038/nn.2334.
Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma. 2000;17(10):811–29.
Tuttolomondo A, Di Sciacca R, Di Raimondo D, Arnao V, Renda C, Pinto A, et al. Neuron protection as a therapeutic target in acute ischemic stroke. Curr Top Med Chem. 2009;9(14):1317–34.
Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, et al. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ Res. 1989;65(3):607–22.
Khalid MA, Ashraf M. Direct detection of endogenous hydroxyl radical production in cultured adult cardiomyocytes during anoxia and reoxygenation. Is the hydroxyl radical really the most damaging radical species? Circ Res. 1993;72(4):725–36.
Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet. 1994;344(8924):721–4.
Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5(5):597–607. doi:10.1089/152308603770310266.
Huang Y, Rabb H, Womer KL. Ischemia–reperfusion and immediate T cell responses. Cell Immunol. 2007;248(1):4–11. doi:10.1016/j.cellimm.2007.03.009.
Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, et al. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41(2–3):172–9. doi:10.1007/s12035-010-8102-z.
Lv M, Liu Y, Zhang J, Sun L, Liu Z, Zhang S, et al. Roles of inflammation response in microglia cell through Toll-like receptors 2/interleukin-23/interleukin-17 pathway in cerebral ischemia/reperfusion injury. Neuroscience. 2011;176:162–72. doi:10.1016/j.neuroscience.2010.11.066.
Chaudhry K, Rogers R, Guo M, Lai Q, Goel G, Liebelt B, et al. Matrix metalloproteinase-9 (MMP-9) expression and extracellular signal-regulated kinase 1 and 2 (ERK1/2) activation in exercise-reduced neuronal apoptosis after stroke. Neurosci Lett. 2010;474(2):109–14. doi:10.1016/j.neulet.2010.03.020.
Curry A, Guo M, Patel R, Liebelt B, Sprague S, Lai Q, et al. Exercise pre-conditioning reduces brain inflammation in stroke via tumor necrosis factor-alpha, extracellular signal-regulated kinase 1/2 and matrix metalloproteinase-9 activity. Neurol Res. 2010;32(7):756–62. doi:10.1179/174313209x459101.
Davis W, Mahale S, Carranza A, Cox B, Hayes K, Jimenez D, et al. Exercise pre-conditioning ameliorates blood–brain barrier dysfunction in stroke by enhancing basal lamina. Neurol Res. 2007;29(4):382–7. doi:10.1179/016164107x204701.
Ding Y, Li J, Luan X, Ding YH, Lai Q, Rafols JA, et al. Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience. 2004;124(3):583–91. doi:10.1016/j.neuroscience.2003.12.029.
Dornbos 3rd D, Ding Y. Mechanisms of neuronal damage and neuroprotection underlying ischemia/reperfusion injury after physical exercise. Curr Drug Targets. 2012;13(2):247–62.
Bramlett HM, Green EJ, Dietrich WD, Busto R, Globus MY, Ginsberg MD. Posttraumatic brain hypothermia provides protection from sensorimotor and cognitive behavioral deficits. J Neurotrauma. 1995;12(3):289–98.
Dietrich WD, Alonso O, Busto R, Globus MY, Ginsberg MD. Post-traumatic brain hypothermia reduces histopathological damage following concussive brain injury in the rat. Acta Neuropathol. 1994;87(3):250–8.
Dietrich WD, Bramlett HM. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics. 2010;7(1):43–50. doi:10.1016/j.nurt.2009.10.015.
Jia F, Mao Q, Liang YM, Jiang JY. Effect of post-traumatic mild hypothermia on hippocampal cell death after traumatic brain injury in rats. J Neurotrauma. 2009;26(2):243–52. doi:10.1089/neu.2008.0670.
Okauchi M, Kawai N, Nakamura T, Kawanishi M, Nagao S. Effects of mild hypothermia and alkalizing agents on brain injuries in rats with acute subdural hematomas. J Neurotrauma. 2002;19(6):741–51. doi:10.1089/08977150260139110.
Karibe H, Zarow GJ, Graham SH, Weinstein PR. Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood–brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1994;14(4):620–7. doi:10.1038/jcbfm.1994.77.
Kawai N, Nakamura T, Okauchi M, Nagao S. Effects of hypothermia on intracranial pressure and brain edema formation: studies in a rat acute subdural hematoma model. J Neurotrauma. 2000;17(3):193–202.
Alkhulaifi AM, Pugsley WB, Yellon DM. The influence of the time period between preconditioning ischemia and prolonged ischemia on myocardial protection. Cardioscience. 1993;4(3):163–9.
Kato H, Araki T, Murase K, Kogure K. Induction of tolerance to ischemia: alterations in second-messenger systems in the gerbil hippocampus. Brain Res Bull. 1992;29(5):559–65.
Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528(1):21–4.
Lin B, Dietrich WD, Ginsberg MD, Globus MY, Busto R. MK-801 (dizocilpine) protects the brain from repeated normothermic global ischemic insults in the rat. J Cereb Blood Flow Metab. 1993;13(6):925–32. doi:10.1038/jcbfm.1993.115.
Lin B, Globus MY, Dietrich WD, Busto R, Martinez E, Ginsberg MD. Differing neurochemical and morphological sequelae of global ischemia: comparison of single- and multiple-insult paradigms. J Neurochem. 1992;59(6):2213–23.
Costa T, Constantino LC, Mendonca BP, Pereira JG, Herculano B, Tasca CI, et al. N-methyl-d-aspartate preconditioning improves short-term motor deficits outcome after mild traumatic brain injury in mice. J Neurosci Res. 2010;88(6):1329–37. doi:10.1002/jnr.22300.
Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, et al. Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J Neurosci Res. 2005;82(5):665–73. doi:10.1002/jnr.20674.
DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab. 2009;29(2):375–84. doi:10.1038/jcbfm.2008.126.
Fanne RA, Nassar T, Mazuz A, Waked O, Heyman SN, Hijazi N, et al. Neuroprotection by glucagon: role of gluconeogenesis. J Neurosurg. 2011;114(1):85–91. doi:10.3171/2010.4.jns10263.
Umschwief G, Shein NA, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E. Heat acclimation provides sustained improvement in functional recovery and attenuates apoptosis after traumatic brain injury. J Cereb Blood Flow Metab. 2010;30(3):616–27. doi:10.1038/jcbfm.2009.234.
Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8(2):101–5.
Yan EB, Hellewell SC, Bellander BM, Agyapomaa DA, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J Neuroinflammation. 2011;8:147. doi:10.1186/1742-2094-8-147.
Wang YC, Lin S, Yang QW. Toll-like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation. 2011;8:134. doi:10.1186/1742-2094-8-134.
Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32(7):1362–76. doi:10.1038/jcbfm.2012.32.
Ravati A, Ahlemeyer B, Becker A, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species. Brain Res. 2000;866(1–2):23–32.
Zhang B, Huang Y, Su Z, Wang S, Wang J, Wang A, et al. Neurological, functional, and biomechanical characteristics after high-velocity behind armor blunt trauma of the spine. J Trauma. 2011;71(6):1680–8. doi:10.1097/TA.0b013e318231bce7.
Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev. 2008;60(13–14):1471–7. doi:10.1016/j.addr.2008.03.020.
Saugstad JA. MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. 2010;30(9):1564–76. doi:10.1038/jcbfm.2010.101.
Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of K(ATP) channels: reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997;87(2):361–70.
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, et al. Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol—a clinical trial. Acta Anaesthesiol Scand. 2012;56(1):30–8. doi:10.1111/j.1399-6576.2011.02585.x.
Bickler PE, Zhan X, Fahlman CS. Isoflurane preconditions hippocampal neurons against oxygen-glucose deprivation: role of intracellular Ca2+ and mitogen-activated protein kinase signaling. Anesthesiology. 2005;103(3):532–9.
Kapinya KJ, Lowl D, Futterer C, Maurer M, Waschke KF, Isaev NK, et al. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke. 2002;33(7):1889–98.
Kawano T, Kunz A, Abe T, Girouard H, Anrather J, Zhou P, et al. iNOS-derived NO and nox2-derived superoxide confer tolerance to excitotoxic brain injury through peroxynitrite. J Cereb Blood Flow Metab. 2007;27(8):1453–62. doi:10.1038/sj.jcbfm.9600449.
Luo Y, Ma D, Ieong E, Sanders RD, Yu B, Hossain M, et al. Xenon and sevoflurane protect against brain injury in a neonatal asphyxia model. Anesthesiology. 2008;109(5):782–9. doi:10.1097/ALN.0b013e3181895f88.
Zhang JH, Lo T, Mychaskiw G, Colohan A. Mechanisms of hyperbaric oxygen and neuroprotection in stroke. Pathophysiology. 2005;12(1):63–77. doi:10.1016/j.pathophys.2005.01.003.
Hu S, Li F, Luo H, Xia Y, Zhang J, Hu R, et al. Amelioration of rCBF and PbtO2 following TBI at high altitude by hyperbaric oxygen pre-conditioning. Neurol Res. 2010;32(2):173–8. doi:10.1179/174313209x414524.
Jadhav V, Ostrowski RP, Tong W, Matus B, Jesunathadas R, Zhang JH. Cyclo-oxygenase-2 mediates hyperbaric oxygen preconditioning-induced neuroprotection in the mouse model of surgical brain injury. Stroke. 2009;40(9):3139–42. doi:10.1161/strokeaha.109.549774.
Kinni H, Guo M, Ding JY, Konakondla S, Dornbos 3rd D, Tran R, et al. Cerebral metabolism after forced or voluntary physical exercise. Brain Res. 2011;1388:48–55. doi:10.1016/j.brainres.2011.02.076.
Liebelt B, Papapetrou P, Ali A, Guo M, Ji X, Peng C, et al. Exercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70 (heat shock protein-72) and extracellular-signal-regulated-kinase 1/2. Neuroscience. 2010;166(4):1091–100. doi:10.1016/j.neuroscience.2009.12.067.
Ding YH, Luan XD, Li J, Rafols JA, Guthinkonda M, Diaz FG, et al. Exercise-induced overexpression of angiogenic factors and reduction of ischemia/reperfusion injury in stroke. Curr Neurovasc Res. 2004;1(5):411–20.
Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009;218(2):363–70. doi:10.1016/j.expneurol.2009.05.026.
Acknowledgment
The part of this work was supported by funds from NINDS RO1 NS 042133 and the Miami Project to Cure Paralysis. The corresponding author (S.Y.) was also partially supported by The General Insurance Association of Japan.
Conflict of Interest
We have no conflict of interest for this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yokobori, S., Mazzeo, A.T., Hosein, K. et al. Preconditioning for Traumatic Brain Injury. Transl. Stroke Res. 4, 25–39 (2013). https://doi.org/10.1007/s12975-012-0226-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-012-0226-1