
Abstract
Carbonic anhydrase 1 (CA-1) is a metalloenzyme present at high concentrations in erythrocytes. Our previous studies showed that erythrocyte lysis contributes to brain edema formation after intracerebral hemorrhage (ICH), and a recent study indicates that CA-1 can cause blood–brain barrier disruption. The present study investigated the role of CA-1 in ICH-induced brain injury. There were three groups in the study. In the first, adult male Sprague Dawley rats received 100 μl autologous blood injection into the right caudate. Sham rats had a needle insertion. Rat brains were used for brain CA-1 level determination. In the second group, rats received an intracaudate injection of either 50 μl CA-1 (1 μg/μl) or saline. Brain water content, microglia activation, and neuronal death (Fluoro-Jade C staining) were examined 24 h later. In the third group, acetazolamide (AZA, 5 μl, 1 mM), an inhibitor of carbonic anhydrases, or vehicle was co-injected with 100 μl blood. Brain water content, neuronal death, and behavioral deficits were measured. We found that CA-I levels were elevated in the ipsilateral basal ganglia at 24 h after ICH. Intracaudate injection of CA-1 induced brain edema (79.0 ± 0.6 vs. 78.0 ± 0.2% in the saline group, p < 0.01), microglia activation, and neuronal death (p < 0.01) at 24 h. AZA, an inhibitor of CA, reduced ICH-induced brain water content (79.3 ± 0.7 vs. 81.0 ± 1.0% in the vehicle-treated group, p < 0.05), neuronal death, and improved functional outcome (p < 0.05). These results suggest that CA-1 from erythrocyte lysis contributes to brain injury after ICH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
After an intracerebral hemorrhage (ICH), lysis of red blood cells (RBCs) in the hematoma results in brain edema formation, neuronal death, and neurological deficits [1–3]. Our previous studies demonstrated that the release of hemoglobin and its degradation products including iron from RBCs are involved in brain injury following ICH [1, 3–6]. However, whether there may be other RBC components that contribute to brain injury has not been investigated.
Carbonic anhydrases are zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate. Carbonic anhydrase I (CA-1), 1 of 14 CA isozymes, is present at high concentrations in RBCs. A recent study found that extracellular CA-1 contributes to hemorrhagic retinal injury and blood–brain barrier disruption [7], but it is not clear whether CA-1 is involved in brain damage after ICH.
The current study investigated: (1) whether CA-1 levels in the perihematomal area are increased after experimental ICH, (2) whether intracerebral injection of CA-1 can cause brain damage, and (3) whether acetazolamide (AZA), a CA inhibitor, reduces brain injury after ICH.
Materials and Methods
Animal Preparation and Intracerebral Blood Infusion
The University of Michigan Committee on the Use and Care of Animals approved animal use protocols. A total of 66 male Sprague Dawley rats weighing 275–300 g (Charles River Laboratories, Portage, MI) were used in the experiments. The animals were anesthetized with pentobarbital (45 mg/kg, i.p.). The right femoral artery was catheterized for blood pressure monitoring, to sample blood for intracerebral infusion, and for analysis of pH, PaO2, PaCO2, hematocrit, and glucose levels. Animal core temperature was maintained at 37°C with the use of a feedback-controlled heating pad. The rats were positioned in a stereotactic frame (Kopf instruments, Tujunga, CA), and a cranial bur hole (1 mm) was drilled on the right coronal suture 3.5 mm lateral to the midline. Autologous whole blood, CA-1, or saline was infused into the right caudate at a rate of 10 μl/min through a 26-gauge needle (coordinates: 0.2 mm anterior, 5.5 mm ventral, and 3.5 mm lateral to the bregma). The needle was removed, the bur hole filled with bone wax, and the skin incision closed with sutures.
Experimental Groups
This study was divided in three parts.
Part I
Rats received an intracaudate injection of 100 μl autologous whole blood. Sham animals had a needle insertion (three rats in each group). The animals were anesthetized again and euthanized 1, 3, and 7 days after ICH. CA-1 levels were measured by Western blot analysis.
Part II
Rats received an intracaudate injection of 50 μl either CA-1 (1 μg/μl) or saline. Some rats were decapitated 24 h later to determine brain water and sodium ion contents (six rats per group). The others were euthanized for histological studies after 24 and 72 h (three rats in each group).
Part III
Rats had an intracaudate injection of 100 μl autologous whole blood with 5 μl AZA (1 mM) or vehicle (4% DMSO). For the measurement of brain water content, rats were killed 24 h later (six rats per group). For brain histological studies, rats were euthanized at 1 and 3 days after ICH (three rats in each group). Behavioral tests were performed in all rats.
Brain Water and Ion Contents
Animals were reanesthetized with pentobarbital (60 mg/kg, i.p.) and brain water and sodium contents measured as previously reported [1]. In brief, the brain was removed quickly, and a 3-mm-thick coronal brain slice was cut 4 mm from the frontal pole. Five tissue samples from each brain were obtained: the ipsilateral and contralateral cortex, the ipsilateral and contralateral basal ganglia, and the cerebellum. Brain samples were immediately weighed on an electronic analytical balance (model AE 100; Mettler Instrument, Highstown, NJ) to obtain the wet weight. Brain samples were then dried at 100°C for 24 h to obtain the dry weight and brain water content calculated as: (wet weight − dry weight)/wet weight × 100%. The dehydrated samples were digested in 1 ml of 1 mol/L nitric acid for 1 week, and the sodium content was measured with an automatic flame photometer (model IL 943; Instrumentation Laboratory, Lexington, MA). Sodium ion content was expressed in milliequivalents per kilogram of dehydrated brain tissue (mEq/kg dry wt).
Western Blot Analysis
Animals were anesthetized and underwent transcardiac perfusion with saline. The brains were removed, and a 3-mm-thick coronal tissue was cut approximately 4 mm from the frontal pole. Brain tissues were sampled 1 mm away from the hematoma to avoid potential red blood cell contamination. The brain tissues were immersed in 0.5 ml Western blot sample buffer and were sonicated for 10 s. Twenty microliters of the sample solution was taken for the protein assay. Western blot analysis was performed as previously reported [8]. Briefly, 50 μg of proteins for each was separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a Hybond-C pure nitrocellulose membrane (Amersham, Piscataway, NJ). The membranes were blocked in nonfat milk and probed with a 1:2,000 dilution of the primary antibody (goat anti-rat CA-1 antibody, Abcam Plc) and a 1:2,000 dilution of a second antibody (peroxidase-conjugated rabbit anti-goat antibody, Jackson Immunoresearch Laboratories Inc.). The antigen/antibody complexes were demonstrated with a chemiluminescence system (Amersham) and exposed to film (X-OMAT; Kodak, Rochester, NY). The relative densities of the bands were analyzed using a National Institute of Health image (version 1.61; NIH, Bethesda, MD).
Fluoro-Jade C Staining
To assess neuronal death, Fluoro-Jade C staining was performed on brain coronal sections [9, 10]. Three high-power images (×40 magnification) were taken around the CA-1 injection site or the hematoma using a digital camera. Fluoro-Jade C-positive cells were counted on these three areas.
Immunohistochemistry
The immunohistochemistry method has been described previously [11]. Briefly, the rats were reanesthetized and perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (pH 7.4). The brains were removed and kept in 4% paraformaldehyde for 6 h, then immersed in 25% sucrose for 3 days at 4°C. After embedding in the mixture of 25% sucrose and OCT (SAKURA Finetek, USA), 18-μm sections were taken on a cryostat. The avidin–biotin complex technique was used in the staining. The primary antibody was mouse anti-rat CD11b (MRC OX42, 1:200 dilution, Serotec Ltd.). Normal mouse serum and the absence of primary antibody were used as negative controls.
Behavioral Tests
Intracerebral hemorrhage-induced neurological deficits were assessed using forelimb placing, forelimb use asymmetry, and corner turn tests [4]. The behavioral tester was blind to the condition of the animal. In the vibrissae-elicited forelimb-placing test, animals were held by their bodies to allow the forelimbs to hang free. Independent testing of each forelimb was conducted by brushing the respective vibrissae on the corner of a table top once per trial for ten trials. A score of one was given each time the rat placed its forelimb onto the edge of the table in response to vibrissae stimulation. The percentage of successful placing responses was determined for the impaired and the unimpaired forelimbs.
For the forelimb use asymmetry test, forelimb use during exploratory activity was analyzed in a standing transparent cylinder. Behavior was quantified by determining the number of times the normal ipsilateral (I) forelimb, the impaired contralateral (C) forelimb, and both (B) forelimbs were used as a percentage of total number of limb usage. A single, overall limb-use asymmetry score was calculated as follows: forelimb use asymmetry score = [I/(I + C + B)] − [C/(I + C + B)].
During the corner turn test, the rat was allowed to proceed into a corner whose angle was 30°. To exit the corner, the animal could turn to either the left or the right, and the direction was recorded. This task was repeated 10 to 15 times, and the percentage of right turns calculated.
Statistical Analysis
All data are presented as mean ± SD. Data were analyzed using the Student t test or an analysis of variance. A probability value of <0.05 was considered statistically significant.
Results
Physiological parameters in all animal groups were measured during intracerebral infusions. Mean arterial blood pressure, blood pH, blood gasses, hematocrit, and blood glucose were controlled within normal ranges.
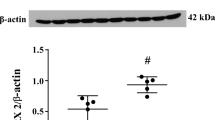
To examine whether CA-1 levels increase in the brain after ICH, rats received an intracaudate injection of 100 μl autologous whole blood. CA-1 levels in the ipsilateral basal ganglia were determined by Western blotting at 1, 3, and 7 days after ICH. We found that CA-1 content in the ipsilateral basal ganglia increased (225 ± 3.3% of the control) at day 1 following hemorrhage (p < 0.05 vs. sham, Fig. 1). CA-1 in the ipsilateral basal ganglia stayed at high levels at day 3 and decreased at day 7 (120.7 ± 33.5% of the control, Fig. 1).

a Western blot analysis showing CA-1 content in the ipsilateral basal ganglia 1 day (lanes 1–3), 3 days (lanes 4–6), and 7 days (lanes 7–9) after 100 μl of blood was injected into the right caudate. Equal amounts of protein (50 μg) were used. CA-1 levels were expressed as percent of the sham control. Values are mean ± SD, *p < 0.05 versus day 7. b CA-1 contents in the ipsilateral (-ipsi) basal ganglia 24 h after sham operation (lanes 1–3) and the ipsilateral basal ganglia (lanes 4–6) and the contralateral (-contra) basal ganglia (lanes 7–9) after ICH. Equal amounts of protein (50 μg) were loaded. CA-1 levels were expressed as percent of the sham control. Values are mean ± SD; #p < 0.01 versus sham and ICH-contra
Whether CA-1 can induce brain edema and cause neuronal death was examined using an intracaudate injection of either CA-1 (50 μg in 50 μl saline) or 50 μl saline. CA-1 injection increased brain water content in the ipsilateral basal ganglia 24 h later (79.0 ± 0.6% versus 78.0 ± 0.2% in the saline group, p < 0.05, Fig. 2), and this edema was associated with sodium ion accumulation (223 ± 31 versus 182 ± 16 mEq/kg dry wt in the saline group, p < 0.05, Fig. 2). Intracaudate injection of CA-1 also caused neuronal death. There were more Fluoro-Jade C-positive cells in the ipsilateral basal ganglia 24 h (196 ± 4 versus 5 ± 4/mm2 in the saline group, p < 0.01) and 72 h (187 ± 22 versus 7 ± 7/mm2 in the saline group, p < 0.01) after CA-1 infusion (Fig. 3). In addition, CA-1 also resulted in microglia activation in the ipsilateral basal ganglia (Fig. 4).

Bar graphs showing brain water (a) and sodium ion (b) contents 24 h after intracaudate injection of CA-1 or saline. Values are mean ± SD; *p < 0.05 versus saline group

Fluoro-Jade C staining showing degenerated neurons in the ipsilateral basal ganglia 1 and 3 days after intracaudate injection of either saline or CA-1. Values are mean ± SD; #p < 0.01 versus saline group

OX-42 immunohistochemistry in the ipsilateral basal ganglia 3 days after intracaudate injection of either saline or CA-1. Values are mean ± SD; #p < 0.01 versus saline group
AZA, an inhibitor of CA, was used to examine the role of CA-1 in ICH-induced brain damage. Intracerebral injection of AZA reduced perihematomal edema (79.3 ± 0.7% versus 81.0 ± 1.0% in the vehicle-treated group, p < 0.05, Fig. 5) and sodium accumulation (249 ± 39 versus 359 ± 80 mEq/kg dry wt, in the vehicle-treated group, p < 0.05, Fig. 5) at 24 h. AZA also reduced neuronal death in the ipsilateral basal ganglia (Fig. 6) and improved forelimb placing, forelimb use asymmetry, and corner turn scores after ICH (Fig. 7).

Bar graphs showing the effects AZA on brain water (a) and sodium ion (b) contents 24 h after ICH. Values are mean ± SD; *p < 0.05 versus the vehicle group

Bar graph showing the effects of AZA on ICH-induced neuronal death. Values are mean ± SD. #p < 0.01 versus the vehicle group

Bar graphs displaying AZA improved behavioral outcome including forelimb placing (a), forelimb use asymmetry (b), and corner turn (c) scores 24 h after ICH. Values are mean ± SD; #p < 0.01 or *p < 0.05 versus the vehicle group
Discussion
The major findings of present study are: (1) CA-1 content is increased in the perihematomal zone after ICH; (2) intracaudate injection of CA-1 results in brain edema, microglia activation, and neuronal death; (3) a CA inhibitor, acetazolamide, reduces brain edema, neuronal death, and neurological deficits after ICH. In combination, these results suggest that CA-1 has a role in brain injury after ICH.
Our previous studies found that components from RBCs cause brain swelling, cell death, and neurological deficits following ICH [1, 12]. For example, hemoglobin and its degradation products, including iron, are neurotoxic. The CA-1 content of RBCs is very high with an average concentration of 12 mg/g hemoglobin, and CA-1 is released after lysis of RBCs from the clot [13]. As a recent study demonstrated that CA-1 causes blood–brain barrier disruption [7], it was important to investigate the role of CA-1 in brain damage following ICH.
The current study indicates that CA-1 contributes to brain edema formation after ICH. Brain edema around the hematoma is commonly observed during the acute and subacute stages following ICH, contributing to poor outcomes [14–16]. Although the mechanisms of edema formation following ICH are not fully resolved, several mechanisms are responsible for edema development. These include hydrostatic pressure during hematoma formation and clot retraction, coagulation cascade activation and thrombin production, hemoglobin and iron toxicity, complement activation, mass effect, secondary perihematomal ischemia, and blood–brain barrier (BBB) disruption [1, 3, 17–22]. The main form of brain edema following ICH is vasogenic edema, resulting from BBB disruption. Although the mechanisms of brain edema formation caused by CA-1 remained to be determined, Gao et al. [7] found that extracellular CA-1 can cause blood-retinal barrier and BBB disruption.
The molecular mechanisms underlying CA-1-induced BBB disruption remain to be fully elucidated. However, Gao et al. found that CA-1 caused kallikrein–kinin activation and that inhibiting that system and bradykinin receptors blocked CA-1-induced blood-retinal barrier disruption [7]. The role of bradykinin in ICH-induced brain injury has not been examined, but there is evidence for a role in brain edema formation and BBB in traumatic brain injury and cerebral ischemia [23–25].
Carbonic anhydrases can elevate pH and increase not only kallikrein enzymatic activity but also factor XIIa activity [7, 26]. However, we do not know the effect of carbonic anhydrase release on brain pH after ICH. Although acidosis occurs in the brain after cerebral ischemia [27], ischemia does not occur following ICH unless the hematoma is massive [3, 28]. We and others have reported a marked increase in perihematomal lactate after ICH [29, 30], but in contrast to ischemia, this does not appear to change brain or CSF pH [29, 31, 32]. While noted by others [29], this dissociation between lactate and pH after ICH is unexplained. It is possible that carbonic anhydrase I contributes to brain lactate and pH dissociation following ICH by producing bicarbonate (deoxyhemoglobin may also contribute by binding H+ ions).
Complement activation contributes to brain edema formation after ICH and may be involved in brain edema caused by CA-1 [20]. Complement is normally excluded from the brain parenchyma by the BBB, but entry can occur after ICH as part of the extravasated blood or later because of BBB disruption. There is evidence that the complement cascade is activated in brain parenchyma after ICH and that it is a potential therapeutic target. For example, N-acetylheparin, a heparin congener without anticoagulant properties, inhibits the complement cascade and attenuates perihematomal brain edema [20]. Interestingly, complement 1 inhibitor attenuates retinal edema induced by CA-1 [7]. Although complement 1 inhibitor also interacts with the kallikrein–kinin system [7], future studies should determine whether or not complement is involved in the brain edema formation caused by CA-1.
CA-1 not only caused brain edema, it also induced neuronal death. Many Fluoro-Jade C-positive cells were found in the ipsilateral basal ganglia 1 and 3 days after CA-1 injection. The mechanisms of CA-1-induced neuronal death are unknown, but evidence showed that carbonic anhydrases regulate postsynaptic Ca2+ influx in hippocampal neurons [33]. A report showed that CA-II-deficient mice are more resistant to hypoxia-induced neuronal damage, which is associated with the alterations of N-methyl-d-aspartate receptors [34]. In addition, blocker of carbonic anhydrase exerted a potent neuroprotection of reducing neuronal apoptosis via pH stabilization [34, 35].
AZA, a CA inhibitor, is an effective drug for preventing high-altitude cerebral edema [36], and it has been used extensively in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) and glaucoma because of its ability to regulate fluid secretion [37, 38]. It has also been used as an adjunct therapy for epilepsy [39]. Our present results demonstrated that treatment with AZA can reduce ICH-induced brain edema, neuronal death, and behavioral deficits suggesting a role of CA in brain damage following ICH and that CA may be a therapeutic target. One brief trial in Romania suggests that AZA has beneficial effects in ICH patients [40]. AZA (usually in combination with furosemide) was used extensively to treat post-hemorrhagic ventricular dilatation in low birth weight children because of its effects on cerebrospinal fluid production, but this was found to be associated with increased neurologic morbidity [41].
Studies found that there are at least 14 known CA isozymes, which participate in a variety of physiological processes including pH homeostasis, fluid secretion, and regulating vascular and glial cell function [42–44]. While AZA is an inhibitor of CA, it has other effects. For example, thrombin plays a key role in brain injury following ICH [45], and AZA can block thrombin-stimulated platelet aggregation [46]. Therefore, the exact mechanisms of AZA-induced brain protection in ICH should be investigated further.
In summary, ICH results in an increase of CA-1 levels in the brain which causes brain damage. Carbonic anhydrase 1 may be a new therapeutic target for ICH.
References
Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–6.
Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002;953(1–2):45–52.
Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral hemorrhage. Lancet Neurol. 2006;5(1):53–63.
Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33(10):2478–84.
Huang F, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96(2):287–93.
Wu G, Xi G, Hua Y, Sagher O. T2* magnetic resonance imaging sequences reflect brain tissue iron deposition following intracerebral hemorrhage. Translat Stroke Res. 2010;1:31–4.
Gao BB, Clermont A, Rook S, Fonda SJ, Srinivasan VJ, Wojtkowski M, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007;13(2):181–8.
Xi G, Keep RF, Hua Y, Xiang JM, Hoff JT. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 1999;30(6):1247–55.
Okauchi M, Xi G, Keep RF, Hua Y. Tissue-type transglutaminase and the effects of cystamine on intracerebral hemorrhage-induced brain edema and neurological deficits. Brain Res. 2009;1249:229–36.
Schmued LC, Albertson C, Slikker Jr W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46.
Gong Y, Hua Y, Keep RF, Hoff JT, Xi G. Intracerebral hemorrhage: effects of aging on brain edema and neurological deficits. Stroke. 2004;35(11):2571–5.
Xi G, Hua Y, Bhasin RR, Ennis SR, Keep RF, Hoff JT. Mechanisms of edema formation after intracerebral hemorrhage: effects of extravasated red blood cells on blood flow and blood–brain barrier integrity. Stroke. 2001;32(12):2932–8.
Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375–401.
Ropper AH, King RB. Intracranial pressure monitoring in comatose patients with cerebral hemorrhage. Arch Neurol. 1984;41(7):725–8.
Ropper AH. Lateral displacement of the brain and level of consciousness in patients with an acute hemispheral mass. N Engl J Med. 1986;314(15):953–8.
Zazulia AR, Diringer MN, Derdeyn CP, Powers WJ. Progression of mass effect after intracerebral hemorrhage. Stroke. 1999;30:1167–73.
Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE, et al. Lobar intracerebral hemorrhage model in pigs: rapid edema development in perihematomal white matter. Stroke. 1996;27(3):490–7.
Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996;84(1):91–6.
Xi G, Wagner KR, Keep RF, Hua Y, de Courten-Myers GM, Broderick JP, et al. The role of blood clot formation on early edema development following experimental intracerebral hemorrhage. Stroke. 1998;29:2580–6.
Hua Y, Xi G, Keep RF, Hoff JT. Complement activation in the brain after experimental intracerebral hemorrhage. J Neurosurg. 2000;92(6):1016–22.
Xi G, Hua Y, Keep RF, Younger JG, Hoff JT. Systemic complement depletion diminishes perihematomal brain edema. Stroke. 2001;32(1):162–7.
Mendelow AD. Mechanisms of ischemic brain damage with intracerebral hemorrhage. Stroke. 1993;24(12 Suppl):I115–7. discussion I8-9.
Maier-Hauff K, Baethmann AJ, Lange M, Schurer L, Unterberg A. The kallikrein-kinin system as mediator in vasogenic brain edema. Part 2: studies on kinin formation in focal and perifocal brain tissue. J Neurosurg. 1984;61(1):97–106.
Zweckberger K, Plesnila N. Anatibant, a selective non-peptide bradykinin B2 receptor antagonist, reduces intracranial hypertension and histopathological damage after experimental traumatic brain injury. Neurosci Lett. 2009;454(2):115–7.
Ding-Zhou L, Margaill I, Palmier B, Pruneau D, Plotkine M, Marchand-Verrecchia C. LF 16–0687 Ms, a bradykinin B2 receptor antagonist, reduces ischemic brain injury in a murine model of transient focal cerebral ischemia. Br J Pharmacol. 2003;139(8):1539–47.
Gardner TW, Antonetti DA. A prize catch for diabetic retinopathy. Nat Med. 2007;13(2):131–2.
Wagner KR, Kleinholz M, de Courten-Myers GM, Myers RE. Hyperglycemic versus normoglycemic stroke: topography of brain metabolites, intracellular pH, and infarct size. J Cereb Blood Flow Metab. 1992;12(2):213–22.
Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373(9675):1632–44.
Mun-Bryce S, Kroh FO, White J, Rosenberg GA. Brain lactate and pH dissociation in edema: 1H- and 31P-NMR in collagenase-induced hemorrhage in rats. Am J Physiol. 1993;265(3 Pt 2):R697–702.
Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE. Early metabolic alterations in edematous perihematomal brain regions following experimental intracerebral hemorrhage. J Neurosurg. 1998;88(6):1058–65.
Fujishima M, Sugi T, Choki J, Yamaguchi T, Omae T. Cerebrospinal fluid and arterial lactate, pyruvate and acid-base balance in patients with intracranial hemorrhages. Stroke. 1975;6(6):707–14.
Sugi T, Fujishima M, Omae T. Lactate and pyruvate concentrations, and acid-base balance of cerebrospinal fluid in experimentally induced intracerebral and subarachnoid hemorrhage in dogs. Stroke. 1975;6(6):715–9.
Fedirko N, Avshalumov M, Rice ME, Chesler M. Regulation of postsynaptic Ca2+ influx in hippocampal CA1 pyramidal neurons via extracellular carbonic anhydrase. J Neurosci. 2007;27(5):1167–75.
Velisek L, Moshe SL, Stanton PK. Resistance of hippocampal synaptic transmission to hypoxia in carbonic anhydrase II-deficient mice. Brain Res. 1995;671(2):245–53.
Kniep EM, Roehlecke C, Ozkucur N, Steinberg A, Reber F, Knels L, et al. Inhibition of apoptosis and reduction of intracellular pH decrease in retinal neural cell cultures by a blocker of carbonic anhydrase. Invest Ophthalmol Vis Sci. 2006;47(3):1185–92.
Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345(2):107–14.
Celebisoy N, Gokcay F, Sirin H, Akyurekli O. Treatment of idiopathic intracranial hypertension: topiramate vs acetazolamide, an open-label study. Acta Neurol Scand. 2007;116(5):322–7.
Lam DS, Lai JS, Tham CC, Chua JK, Poon AS. Argon laser peripheral iridoplasty versus conventional systemic medical therapy in treatment of acute primary angle-closure glaucoma: a prospective, randomized, controlled trial. Ophthalmology. 2002;109(9):1591–6.
Katayama F, Miura H, Takanashi S. Long-term effectiveness and side effects of acetazolamide as an adjunct to other anticonvulsants in the treatment of refractory epilepsies. Brain Dev. 2002;24:150–4.
Popa G, Amaireh M, Dinu M, Jipescu I, Alaicescu M, Stanescu A, et al. Acetazolamide therapy evaluation in haemorrhagic stroke. Rom J Neurol Psychiatry. 1995;33(2):145–55.
Kennedy CR, Ayers S, Campbell MJ, Elbourne D, Hope P, Johnson A. Randomized, controlled trial of acetazolamide and furosemide in posthemorrhagic ventricular dilation in infancy: follow-up at 1 year. Pediatrics. 2001;108(3):597–607.
Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–81.
Pan P, Leppilampi M, Pastorekova S, Pastorek J, Waheed A, Sly WS, et al. Carbonic anhydrase gene expression in CA II-deficient (Car2−/−) and CA IX-deficient (Car9−/−) mice. J Physiol. 2006;571(Pt 2):319–27.
Svichar N, Esquenazi S, Waheed A, Sly WS, Chesler M. Functional demonstration of surface carbonic anhydrase IV activity on rat astrocytes. Glia. 2006;53(3):241–7.
Hua Y, Keep R, Hoff J, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38:759–62.
Siffert W, Fox G, Gros G. The effect of carbonic anhydrase inhibitin on the velocity of thrombin-stimulated platelet aggregation under physiological condition. Biochem Biophys Res Commun. 1984;121(1):266–70.
Acknowledgments
This study was supported by grants NS-017760, NS-039866, and NS-057539 from the National Institutes of Health (NIH) and 0840016N from the American Heart Association (AHA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and AHA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guo, F., Hua, Y., Wang, J. et al. Inhibition of Carbonic Anhydrase Reduces Brain Injury After Intracerebral Hemorrhage. Transl. Stroke Res. 3, 130–137 (2012). https://doi.org/10.1007/s12975-011-0106-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-011-0106-0